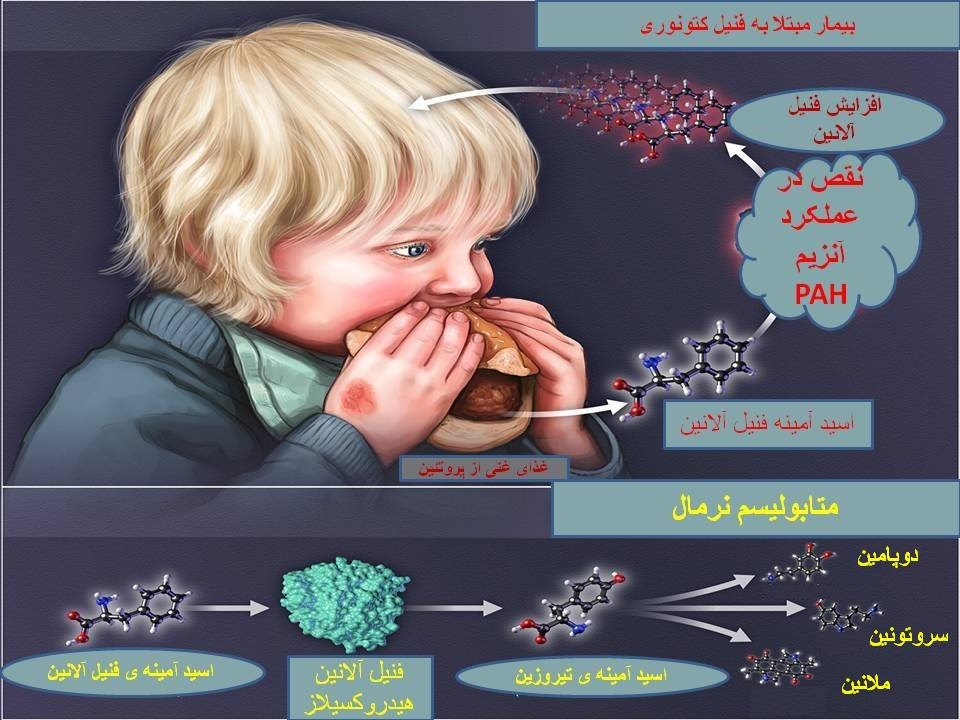
فنیل کتونوری (Phenylketonuria) یا PKU شایعترین بیماری مرتبط با متابولیسم اسید آمینه است. این بیماری گاهی به اسم "کمبود فنیل آلانین هیدروکسیلاز" نیز شناخته میشود. کمبود فنیل آلانین هیدروکسیلاز(PAH) باعث ایجاد اختلال در متابولیسم اسید آمینه ضروری فنیل آلانین میشود و منجر به تجمع این اسیدآمینه در مایعات بدن میگردد. افزایش سطح فنیل آلانین در بدن بر عملکرد مغز تاثیر میگذارد. افراد مبتلا به PKU تقریباً همیشه ناتوانی ذهنی دارند مگر اینکه سطح آن از طریق رژیم غذایی یا درمان دارویی کنترل شود.
بارزترین مشخصه پوستی این بیماران، داشتن پوست و مو روشن میباشد که ناشی از اختلال در سنتز ملانین است. افراد تحت درمان معمولاً رنگدانههای نرمالی دارند. سایر علائم پوستی مرتبط با این بیماری عبارتند از: اگزما، حساسیت به نور، افزایش بروز عفونتهای پیوژنیک، افزایش بروز کراتوز پیلاریس، کاهش تعداد خالهای رنگی، پلاکهای اسکلرودرمی و ریزش مو.
سایر علائم این بیماری در افراد درمان نشده عبارتند از؛ ناتوانی ذهنی (شایعترین علامت)، بوی نامطبوع، صرع، علائمی شبیه بیماری پارکینسون و ناهنجاریهای چشمی.
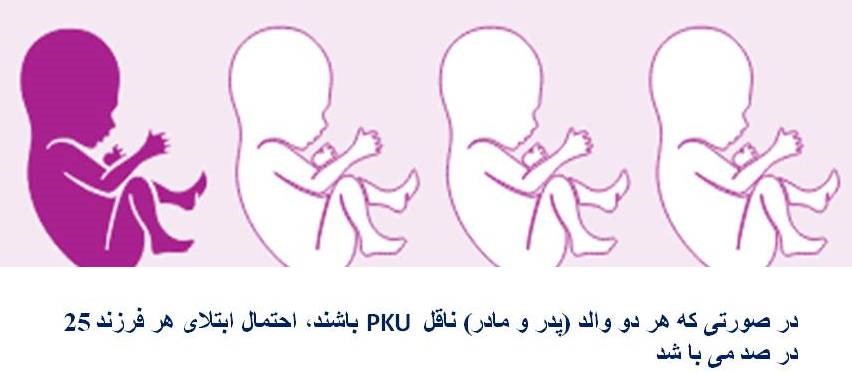
این بیماری معمولاً به صورت اتوزومال مغلوب به ارث میرسد، و این به این معنی است که هر دو کپی ژن در هر سلول دارای جهش هستند. والدین یک فرد مبتلا به بیماری، هر کدام ناقل یک ژن جهش یافته هستند، اما علائم و نشانههای بیماری را نشان نمیدهند. در بیماریهای اتوزومال مغلوب در صورتی که والدین هر دو ناقل باشند، احتمال داشتن فرزند مبتلا در هر بارداری 25 درصد است.
برای تشخیص این بیماری سطح فنیل آلانین تعیین میشود. اسیدهای آمینه پلاسما اندازهگیری میشوند و افزایش فنیل آلانین و کاهش تیروزین مورد توجه قرار میگیرد. همچنین اسیدهای آمینه ادرار و دفع ادراری فنیل آلانین مورد تجزیه و تحلیل قرار گرفته میشود.
مواد دیگری نیز مانند آلفا کتو اسیدها و هیدروکسی اسیدها، هیدروکسی فنیل استات، بیوپترین و ماندلات نیز در آنالیز ادرار بیماران مورد بررسی قرار میگیرند.
امروزه تستهای مولکولی و آنالیز جهشهای ژن PAH نیز به ویژه در موارد تشخیص پیش از تولد (PND) و تشخیص پیش از انتقال جنین به رحم (PGD) مورد استفاده قرار میگیرند.
در اغلب بیماران، فرم کلاسیک بیماری مربوط به نقص درPAH میباشد که منجر به افزایش فنیل آلانین در پلاسما ( بیشتر از 1200 میکرو مول در لیتر، مقادیر نرمال بین 35-90) و افزایش دفع ادراری فنیل پیروویک اسید و فنیل استیک اسید میگردد. فنیل آلانین هیدروکسیلاز تبدیل فنیل آلانین به تیروزین را کاتالیز میکند. مکانیسمی که توسط آن افزایش فنیل آلانین باعث ناتوانی ذهنی میشود شناخته نشده است. اما محدود کردن مصرف فنیل آلانین در دوران نوزادی باعث بهبود وضعیت افراد میشود. بین کنترل سطح فنیل آلانین خون در کودکی وIQ رابطهای محکم وجود دارد. فنیل آلانین هیدروکسیلاز به یک کوفاکتور غیر پروتئینی به نام تتراهیدروبیوپترین (BH4) نیاز دارد. درصد کمی از کودکان با سطح فنیل آلانین بالا، سطح پروتئینPAH طبیعی را نشان میدهند، ولی میزان BH4 پایینی دارند که به عنوان کمبود تتراهیدروبیوپترین شناخته میشود. این وضعیت گاهی اوقات PKU بدخیم نامیده میشود و میتواند ناشی از جهشهای دو آللی در ژنهای GCH1، PCB1، PTS یا QDPR باشد .کوفاکتور BH4 همچنین برای هیدروکسیلاسیون تیروزین (پیش ساز دوپامین) و تریپتوفان (پیش ساز سروتونین) مورد نیاز است. بنابراین، افراد مبتلا به کمبود کوفاکتور BH4 میتوانند مشکلات عصبی بیشتری داشته باشند که به تنهایی با کاهش فنیل آلانین رژیم غذایی به طور کامل اصلاح نمیشوند، اما اغلب به درمانهای اضافی نیاز دارند.
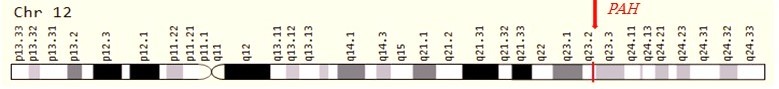
فنیل کتونوری (PKU) یک اختلال اتوزومال مغلوب است که در اثر جهش در ژن PAH ایجاد میشود و محصول پروتئینی آن فنیل آلانین هیدروکسیلاز است. این ژن در 12q23.2 قرار دارد و حدود 171 کیلوبایت باز را شامل میشود که دارای 13 اگزون است. بیش از 500 جهش مختلف در ژن PAH شناسایی شده است. ژن PAH تنوع آللی زیادی را نشان میدهد و جهشهای بیماریزا در تمام 13 اگزون ژن PAH و نواحی بالا دست و پایین دست ژن دیده شدهاند. انواع مختلف جهشها مانندmissense mutation ، splice variant، حذفهای بزرگ و کوچک و ... در ژن PAH گزارش شدهاند.
فراوانی این بیماری در جمعیتهای مختلف متفاوت است. در ایالات متحده شیوع آن 4 مورد در هر 100000 نفر است. در ترکیه و یهودیهای یمن، شیوع بالا و در فنلاند و ژاپن شیوع پایین گزارش شده است. فنیل کتونوری به دلیل غربالگری نوزادان معمولاً در دوران نوزادی تشخیص داده میشود. در جنسیتهای مختلف تفاوتی ندارند و زنان مبتلا در دوران بارداری باید مصرف فنیل آلانین خود را محدود کنند تا از نقایص مادرزادی و ناتوانی ذهنی در نوزادان خود جلوگیری کنند. در ایالات متحده، PKU در سفیدپوستان رایج است و در سرتاسر جهان، PKU در سفیدپوستان و آسیاییها شایعتر است.
به نظر میرسد آسیبهای عصبی ناشی از افزایش فنیل آلانین در بدن، بلافاصله پس از تولد رخ میدهند. مطالعات نشان میدهند که هر 4 هفته تاخیر در شروع درمان باعث کاهش تقریبی 4 واحد در میزان IQ میگردد. اجماع کلی بر این است که درمان، در سالهای اولیه زندگی تاثیر بیشتری نسبت به سالهای بعد دارد و بطور کلی توصیه میشود که برای جلوگیری از آسیبهای عصبی بهتر است درمان در اسرع وقت شروع شود. شروع درمان قبل از 10 روزگی نتایج بسیار بهتری را در پی دارد. بیماران با غلظت فنیل آلانین بالاتر از 600 میکرومول در لیتر، باید تحت درمان قرار گیرند. در افرادی که میزان فنیل آلانین کمتر از 360 میکرومول بر لیتر داشته باشند بهتر است در یکسال اول زندگی تحت پایش قرار گرفته شوند. در مورد شروع درمان افراد دارای فنیل آلانین بین 360-600 میکرومول در لیتر نظرات متناقضی وجود دارد و ظاهراً افرادی که فنیل آلانین بالاتری دارند، ضریب هوشی پایین تری نیز دارند. پیش بینی می شود به ازای هر 100 واحد افزایش در میزان فنیل آلانین، ضریب هوشی به میزان 6 واحد کاهش یابد.
این ژن در انسان بر روی کروموزوم 12 قرار دارد و دارای 13 اگزون میباشد. و بیش از90 kb طول دارد. تاکنون بیش از 1000 جهش مختلف در بیماران PKU گزارش شدهاست. بیش از 95 درصد این جهشها با روش توالییابی ژن قابل تشخیص هستند. سایر جهشها با روشهای دیگری مانند MLPA و Long-range PCR قابل شناسایی هستند. ژن PAH مسئول سنتز آنزیم فنیل آلانین هیدروکسیلاز میباشد. این آنزیم نقش اساسی در کاتابولیسم اسید آمینه فنیل آلانین دارد. فنیل آلانین در تمام پروتئینها و نیز برخی از شیرینکنندههای مصنوعی یافت میشود. آنزیم فنیل آلانین هیدروکسیلاز تبدیل فنیل آلانین به تیروزین را بر عهده دارد. این آنزیم برای فعالیت خود به تتراهیدروبیوپترین (BH4) نیاز دارد. تیروزین برای ساخت تعدادی از هورمونها، انتقال دهندههای عصبی و ملانین استفاده میشود. تیروزین همچنین میتواند به مولکولهای کوچکتر تبدیل شده و در تولید انرژی نقش داشته باشد. همانطور که قبلاً گفته شد مقادیر بالای فنیل آلانین برای بدن مضر میباشد.
شرکت نماژن آزما مفتخر است تا به عنوان یک شرکت دانشبنیان با اتکا به دانش بینالمللی و کارشناسان خبره در مسیر پیشرفت و بومیسازی فناوریهای علوم زیستی قدم برداشته است.
2025 تمامی حقوق مادی و معنوی این سایت متعلق به مجموعه نماژن می باشد