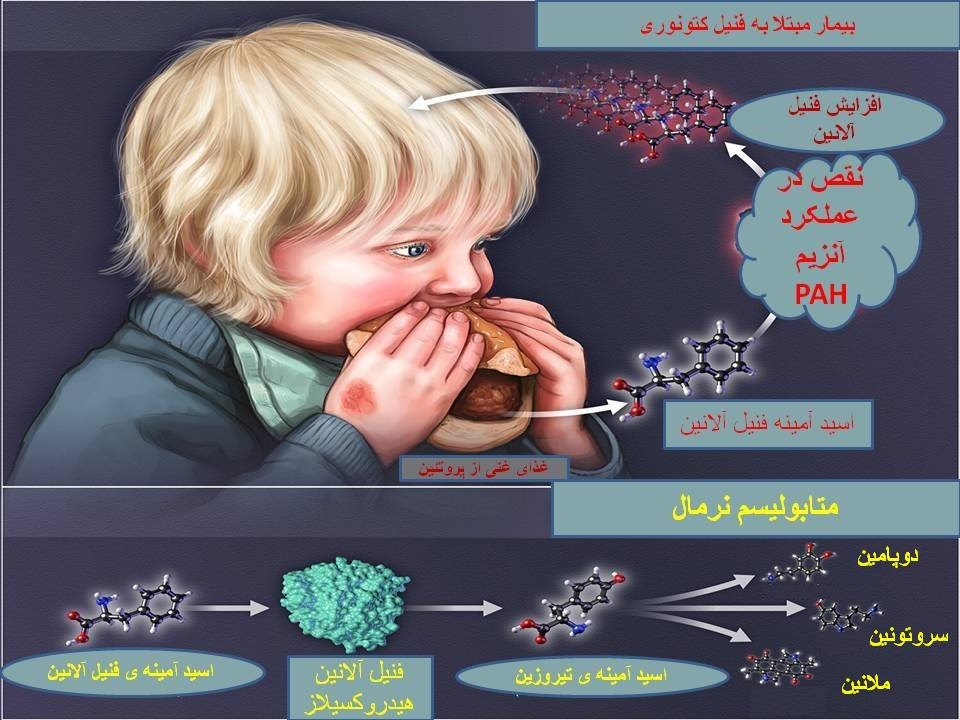
فنیل کتونوری (Phenylketonuria) یا PKU شایعترین بیماری مرتبط با متابولیسم اسید آمینه است. این بیماری گاهی به اسم “کمبود فنیل آلانین هیدروکسیلاز” نیز شناخته میشود. کمبود فنیل آلانین هیدروکسیلاز(PAH) باعث ایجاد اختلال در متابولیسم اسید آمینه ضروری فنیل آلانین میشود و منجر به تجمع این اسیدآمینه در مایعات بدن میگردد. افزایش سطح فنیل آلانین در بدن بر عملکرد مغز تاثیر میگذارد. افراد مبتلا به PKU تقریباً همیشه ناتوانی ذهنی دارند مگر اینکه سطح آن از طریق رژیم غذایی یا درمان دارویی کنترل شود.
بارزترین مشخصه پوستی این بیماران، داشتن پوست و مو روشن میباشد که ناشی از اختلال در سنتز ملانین است. افراد تحت درمان معمولاً رنگدانههای نرمالی دارند. سایر علائم پوستی مرتبط با این بیماری عبارتند از: اگزما، حساسیت به نور، افزایش بروز عفونتهای پیوژنیک، افزایش بروز کراتوز پیلاریس، کاهش تعداد خالهای رنگی، پلاکهای اسکلرودرمی و ریزش مو.
سایر علائم این بیماری در افراد درمان نشده عبارتند از؛ ناتوانی ذهنی (شایعترین علامت)، بوی نامطبوع، صرع، علائمی شبیه بیماری پارکینسون و ناهنجاریهای چشمی.
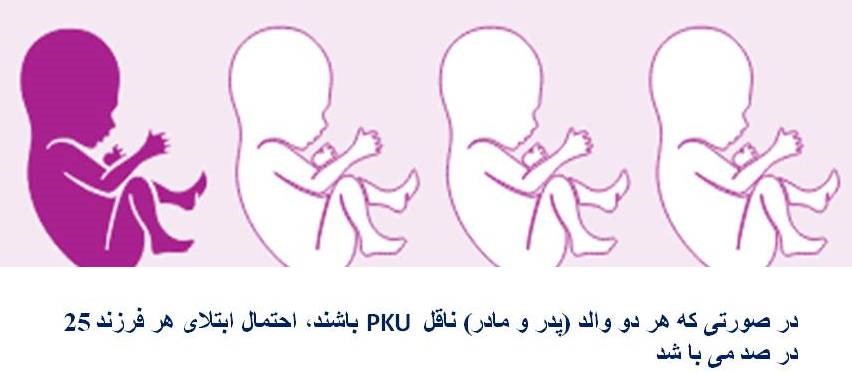
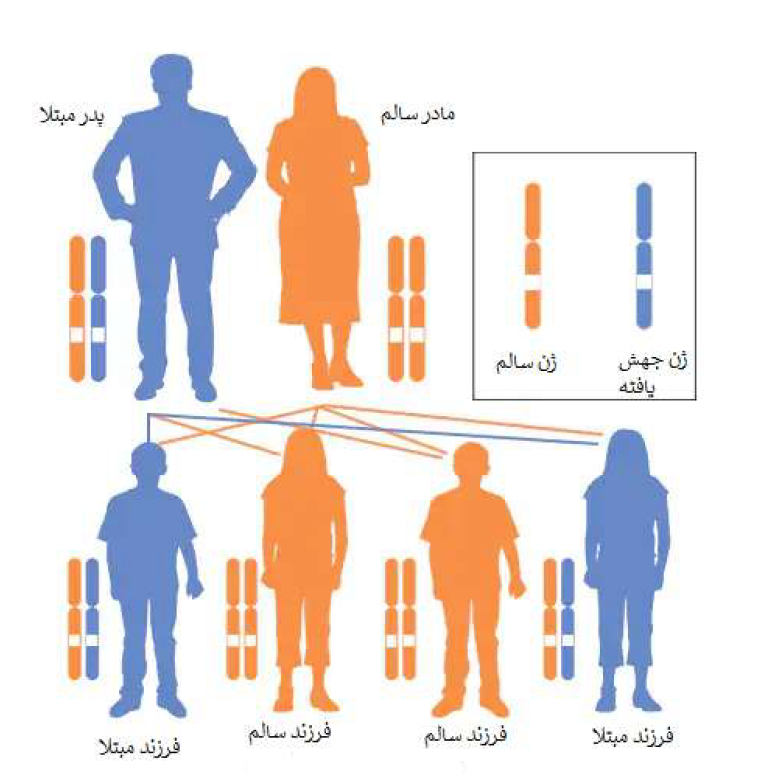
این بیماری معمولاً به صورت اتوزومال مغلوب به ارث میرسد، و این به این معنی است که هر دو کپی ژن در هر سلول دارای جهش هستند. والدین یک فرد مبتلا به بیماری، هر کدام ناقل یک ژن جهش یافته هستند، اما علائم و نشانههای بیماری را نشان نمیدهند. در بیماریهای اتوزومال مغلوب در صورتی که والدین هر دو ناقل باشند، احتمال داشتن فرزند مبتلا در هر بارداری 25 درصد است.
برای تشخیص این بیماری سطح فنیل آلانین تعیین میشود. اسیدهای آمینه پلاسما اندازهگیری میشوند و افزایش فنیل آلانین و کاهش تیروزین مورد توجه قرار میگیرد. همچنین اسیدهای آمینه ادرار و دفع ادراری فنیل آلانین مورد تجزیه و تحلیل قرار گرفته میشود.
مواد دیگری نیز مانند آلفا کتو اسیدها و هیدروکسی اسیدها، هیدروکسی فنیل استات، بیوپترین و ماندلات نیز در آنالیز ادرار بیماران مورد بررسی قرار میگیرند.
امروزه تستهای مولکولی و آنالیز جهشهای ژن PAH نیز به ویژه در موارد تشخیص پیش از تولد (PND) و تشخیص پیش از انتقال جنین به رحم (PGD) مورد استفاده قرار میگیرند.
در اغلب بیماران، فرم کلاسیک بیماری مربوط به نقص درPAH میباشد که منجر به افزایش فنیل آلانین در پلاسما ( بیشتر از 1200 میکرو مول در لیتر، مقادیر نرمال بین 35-90) و افزایش دفع ادراری فنیل پیروویک اسید و فنیل استیک اسید میگردد. فنیل آلانین هیدروکسیلاز تبدیل فنیل آلانین به تیروزین را کاتالیز میکند. مکانیسمی که توسط آن افزایش فنیل آلانین باعث ناتوانی ذهنی میشود شناخته نشده است. اما محدود کردن مصرف فنیل آلانین در دوران نوزادی باعث بهبود وضعیت افراد میشود. بین کنترل سطح فنیل آلانین خون در کودکی وIQ رابطهای محکم وجود دارد. فنیل آلانین هیدروکسیلاز به یک کوفاکتور غیر پروتئینی به نام تتراهیدروبیوپترین (BH4) نیاز دارد. درصد کمی از کودکان با سطح فنیل آلانین بالا، سطح پروتئینPAH طبیعی را نشان میدهند، ولی میزان BH4 پایینی دارند که به عنوان کمبود تتراهیدروبیوپترین شناخته میشود. این وضعیت گاهی اوقات PKU بدخیم نامیده میشود و میتواند ناشی از جهشهای دو آللی در ژنهای GCH1، PCB1، PTS یا QDPR باشد .کوفاکتور BH4 همچنین برای هیدروکسیلاسیون تیروزین (پیش ساز دوپامین) و تریپتوفان (پیش ساز سروتونین) مورد نیاز است. بنابراین، افراد مبتلا به کمبود کوفاکتور BH4 میتوانند مشکلات عصبی بیشتری داشته باشند که به تنهایی با کاهش فنیل آلانین رژیم غذایی به طور کامل اصلاح نمیشوند، اما اغلب به درمانهای اضافی نیاز دارند.
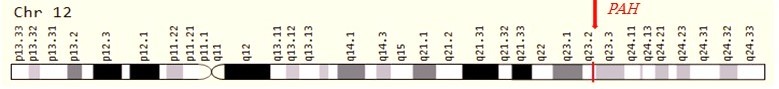
فنیل کتونوری (PKU) یک اختلال اتوزومال مغلوب است که در اثر جهش در ژن PAH ایجاد میشود و محصول پروتئینی آن فنیل آلانین هیدروکسیلاز است. این ژن در 12q23.2 قرار دارد و حدود 171 کیلوبایت باز را شامل میشود که دارای 13 اگزون است. بیش از 500 جهش مختلف در ژن PAH شناسایی شده است. ژن PAH تنوع آللی زیادی را نشان میدهد و جهشهای بیماریزا در تمام 13 اگزون ژن PAH و نواحی بالا دست و پایین دست ژن دیده شدهاند. انواع مختلف جهشها مانندmissense mutation ، splice variant، حذفهای بزرگ و کوچک و … در ژن PAH گزارش شدهاند.
فراوانی این بیماری در جمعیتهای مختلف متفاوت است. در ایالات متحده شیوع آن 4 مورد در هر 100000 نفر است. در ترکیه و یهودیهای یمن، شیوع بالا و در فنلاند و ژاپن شیوع پایین گزارش شده است. فنیل کتونوری به دلیل غربالگری نوزادان معمولاً در دوران نوزادی تشخیص داده میشود. در جنسیتهای مختلف تفاوتی ندارند و زنان مبتلا در دوران بارداری باید مصرف فنیل آلانین خود را محدود کنند تا از نقایص مادرزادی و ناتوانی ذهنی در نوزادان خود جلوگیری کنند. در ایالات متحده، PKU در سفیدپوستان رایج است و در سرتاسر جهان، PKU در سفیدپوستان و آسیاییها شایعتر است.
به نظر میرسد آسیبهای عصبی ناشی از افزایش فنیل آلانین در بدن، بلافاصله پس از تولد رخ میدهند. مطالعات نشان میدهند که هر 4 هفته تاخیر در شروع درمان باعث کاهش تقریبی 4 واحد در میزان IQ میگردد. اجماع کلی بر این است که درمان، در سالهای اولیه زندگی تاثیر بیشتری نسبت به سالهای بعد دارد و بطور کلی توصیه میشود که برای جلوگیری از آسیبهای عصبی بهتر است درمان در اسرع وقت شروع شود. شروع درمان قبل از 10 روزگی نتایج بسیار بهتری را در پی دارد. بیماران با غلظت فنیل آلانین بالاتر از 600 میکرومول در لیتر، باید تحت درمان قرار گیرند. در افرادی که میزان فنیل آلانین کمتر از 360 میکرومول بر لیتر داشته باشند بهتر است در یکسال اول زندگی تحت پایش قرار گرفته شوند. در مورد شروع درمان افراد دارای فنیل آلانین بین 360-600 میکرومول در لیتر نظرات متناقضی وجود دارد و ظاهراً افرادی که فنیل آلانین بالاتری دارند، ضریب هوشی پایین تری نیز دارند. پیش بینی می شود به ازای هر 100 واحد افزایش در میزان فنیل آلانین، ضریب هوشی به میزان 6 واحد کاهش یابد.
این ژن در انسان بر روی کروموزوم 12 قرار دارد و دارای 13 اگزون میباشد. و بیش از90 kb طول دارد. تاکنون بیش از 1000 جهش مختلف در بیماران PKU گزارش شدهاست. بیش از 95 درصد این جهشها با روش توالییابی ژن قابل تشخیص هستند. سایر جهشها با روشهای دیگری مانند MLPA و Long-range PCR قابل شناسایی هستند. ژن PAH مسئول سنتز آنزیم فنیل آلانین هیدروکسیلاز میباشد. این آنزیم نقش اساسی در کاتابولیسم اسید آمینه فنیل آلانین دارد. فنیل آلانین در تمام پروتئینها و نیز برخی از شیرینکنندههای مصنوعی یافت میشود. آنزیم فنیل آلانین هیدروکسیلاز تبدیل فنیل آلانین به تیروزین را بر عهده دارد. این آنزیم برای فعالیت خود به تتراهیدروبیوپترین (BH4) نیاز دارد. تیروزین برای ساخت تعدادی از هورمونها، انتقال دهندههای عصبی و ملانین استفاده میشود. تیروزین همچنین میتواند به مولکولهای کوچکتر تبدیل شده و در تولید انرژی نقش داشته باشد. همانطور که قبلاً گفته شد مقادیر بالای فنیل آلانین برای بدن مضر میباشد.
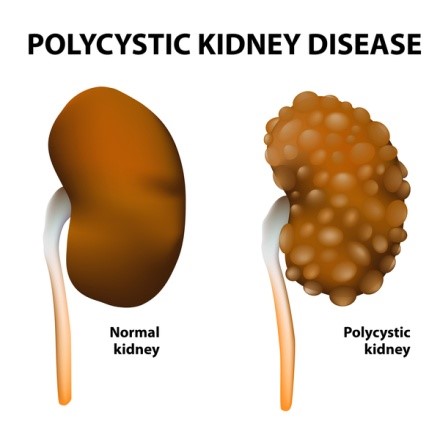
بیماری کلیهی پلیکیستیک عموماً باعث اختلال در عملکرد کلیهها میگردد. در این بیماری کیسههایی پر از مایع، که کیست نامیده میشوند در کلیه تشکیل میشود. وجود این کیستها عملکرد کلیه در تصفیه خون از مواد زائد را مختل میکند. رشد کیستها باعث بزرگتر شدن کلیهها میگردد. این کیستها علاوه بر کلیهها ممکن است در سایر اندامها بهویژه کبد نیز وجود داشته باشند.
مهمترین علائم بیماری کلیهی پلیکیستیک شامل: فشار خون بالا، درد در پشت یا پهلوها، وجود خون در ادرار، عفونتهای مکرر مجاری ادراری، سنگ کلیه و ناهنجاریهای قلبی میباشد. علاوه بر این افراد مبتلا به بیماری کلیهی پلیکیستیک در معرض ابتلا به آنوریسم آئورت و یا آنوریسم عروق مغزی میباشند. ( آنوریسم نوعی برآمدگی غیر طبیعی در رگهای خونی است که در صورت پاره شدن تهدید کننده حیات فرد میباشد.)
بیماری کلیهی پلیکیستیک به دو شکل اصلی دیده میشود و تفاوت آنها در سن شروع بیماری و نحوه توارث آنها میباشد. در فرم اتوزومال غالب، که ADPKD نیز نامیده میشود، نشانههای بیماری معمولاً در بزرگسالی دیده میشوند، هرچند ممکن است از بدو تولد یا کودکی، کیستهایی در کلیه وجود داشته باشد. بسته به علت ژنتیکی بیماری، شکل اتوزومال غالب بیماری به نوع یک و دو تقسیمبندی میشود. شکل اتوزومال مغلوب بیماری کلیهی پلیکیستیک بسیار نادرتر است و اغلب در اوایل زندگی کشنده است. علایم و نشانههای این نوع از بیماری معمولاً در بدو تولد یا در اوایل نوزادی رویت میشوند.
بیماری کلیهی پلیکیستیک یک اختلال ژنتیک نسبتاً شایع است. در ایالات متحده آمریکا یک نفر در هر 500 تا 1000 نفر به این بیماری مبتلا میباشد. نوع اتوزومال مغلوب شیوع کمتری دارد.
جهش در ژنهای PKD1 ، PKD2 و PKHD1 باعث بیماری کلیهی پلیکیستیک میشود. جهش در ژنهای PKD1 و PKD2 معمولاً نوع اتوزومال غالب این بیماری را ایجاد میکنند و به ترتیب باعث ایجاد ADPKD نوع 1 و نوع 2 میشوند. عملکرد محصول پروتئینی این ژنها، بهطور کامل شناخته نشدهاند. ولی احتمالاً در انتقال سیگنال از خارج سلول به داخل هسته نقش دارند. برای عملکرد طبیعی کلیه، عملکرد این دو پروتئین با هم حائز اهمیت میباشد. جهش در ژن PKD1 یا PKD2 باعث تشکیل هزاران کیست میشود که عملکرد کلیه را مختل میکند. افراد دارای جهش در ژن PKD2 ، به ویژه زنان، معمولاً نسبت به افراد دارای جهش در ژن PKD1 فرم خفیفتر بیماری را دارند. جهش در ژن PKHD1 باعث بیماری کلیهی پلیکیستیک از نوع اتوزومال مغلوب میشود. نقش احتمالی پروتئین حاصل از این ژن نیز انتقال سیگنالهای شیمیایی از خارج سلول به هسته میباشد. در صد بسیار کمی از موارد بیماری کلیهی پلیکیستیک منشا ژنتیکی ندارند و به این موارد کلیهی پلیکیستیک اکتسابی گفته میشود.
این ژن پروتئینی به نام پلی سیستین 1 را کد میکند. این پروتئین دارای یک ناحیه بزرگ ان ترمینال خارج سلولی، نواحی گذرنده از غشاء متعدد و یک ناحیه سی ترمینال سیتوپلاسمی است. پلی سیستین یک پروتئین اینتگرال است و در تنظیم غشایی کاتیون کلسیم و هموستاز کلسیم داخل سلولی نقش دارد. این پروتئین در نمو توبولار کلیه نقش دارد و جهش در این ژن باعث ایجاد کلیهی پلیکیستیک اتوزومال غالب نوع یک میشود. این ژن دارای شش سودوژن میباشد.
پروتئین حاصل از این ژن، پلی سیستین 2 نام دارد و قبل از تولد در کلیهها و در بالغین در بافتهای مختلفی دیده میشود. این پروتئین احتمالاً بهعنوان یک کانال در غشای سلولهای کلیه عمل میکند و انتقال کاتیونها بهویژه یونهای کلسیم را به داخل برعهده دارد.
این ژن، پروتئینی به نام فیبروسیستین یا پلیداکتین را تولید میکند که در سلولهای کلیوی جنینی و بالغین دیده میشود و به میزان کمتری در سلولهای کبد و پانکراس نیز بیان میشود. این پروتئین نیز در انتقال سیگنالهای خارج سلولی به داخل سلول نقش دارد. علاوه بر این ممکن است در اتصال سلولها به یکدیگر و رشد و تقسیم سلولها نیز دخالت داشته باشد.
علاوه بر ژنهای فوق، در سالهای اخیر ژنهای دیگری نیز مورد توجه قرار گرفتهاند که جهشهای موجود در این ژنها نیز میتوانند در ایجاد نوع غالب یا مغلوب بیماری کلیهی پلیکیستیک نقش داشته باشند. تعدادی از این ژنها و نوع توارث آنها در جدول زیر ارائه شدهاند.
| Genes | Associated phenotypes | Inheritance |
|---|---|---|
| DNAJB11 | Autosomal dominant polycystic kidney disease | AD |
| DZIP1L | Polycystic kidney disease 5 | AR |
| GANAB | Polycystic kidney and/or polycystic liver disease 3 | AD |
| HNF1B | Renal cell carcinoma, nonpapillary chromophobe, Renal cysts and diabetes syndrome | AD |
| JAG1 | Alagille syndrome | AD |
| LRP5 | Van Buchem disease, Osteoporosis-pseudoglioma syndrome, Hyperostosis, endosteal, Osteosclerosis, Exudative vitreoretinopathy, Osteopetrosis late-onset form type 1, LRP5 primary osteoporosis | AD/AR/Digenic |
| NOTCH2 | Alagille syndrome, Hajdu-Cheney syndrome | AD |
| PKD1 | Polycystic kidney disease | AD |
| PKD2 | Polycystic kidney disease | AD |
| PKHD1 | Polycystic kidney disease | AR |
| PRKCSH | Polycystic liver disease | AD |
| SEC61A1 | Hyperuricemic nephropathy, familial juvenile 4 | AD |
| SEC63 | Polycystic liver disease | AD |
بیماری کلیهی پلیکیستیک عمدتاً توسط دو ژن PKD1 و PKD2 ایجاد میشوند. در صورت وجود سابقه خانوادگی ADPKD، بهندرت از تستهای ژنتیکی استفاده میشود و معمولاً به تشخیصهای مبتنی بر تصویربرداری بسنده میکنند. علاوه بر این غربالگری جهشهای ژن PKD1 با توجه به بزرگی و پیچیدگی ژن وجود چندین سودوژن همیشه با چالش مواجه بودهاست. در حال حاضر از تکنیکهای مبنتی بر PCR و توالییابی، Next Generation Sequencing(NGS) و در موارد کمتر MLPA برای تشخیص جهشهای مرتبط با بیماری کلیهی پلیکیستیک استفاده میشود. در صورت وجود سابقه خانوادگی از Linkage Analysis نیز میتوان برای تشخیص بیماری استفاده کرد.
MS (Multiple sclerosis) شایعترین بیماری التهابی است که میلینهای اطراف نورونهای سیستم عصبی مرکزی یا CNS را درگیر میکند. MS یک بیماری کاملاً ژنتیکی نیست و فاکتورهای محیطی نیز میتوانند در بروز آن دخیل باشند. افراد مبتلا به MS لزوماً فرزندان بیمار ندارند. همچنین مشخص شده که شیوع این بیماری در زنان دو برابر مردان است. سیستم ایمنی، میلینهای پوشش دهنده اطراف نورونها را مورد حمله قرار داده و منجر به بیماری خودایمنی میشود. در سال 1981 استفاده از تکنولوژی MRI این امکان را فراهم کرد که با تصویربرداری از مغز، بتوان افراد بیمار را حتی بدون هیچ علائم خارجی شناسایی کرد.
MS یک بیماری نسبتا شایع در اروپا، ایالات متحده، کانادا، نیوزلند و بخشهایی از استرالیا میباشد. میزان بروز آن در دوران کودکی کم و بعد از سن 18 سالگی به سرعت افزایش یافته و بین سنین 20 تا 40 سالگی به اوج خود میرسد (در زنان حدود دو سال زودتر از مردان رخ میدهد). پس از آن شیوع بیماری به آرامی کاهش یافته و در افراد 50 سال و بیشتر از 50 سال کمتر دیده میشود. مطالعات نشان داده وقوع MS در زنان نسبت به مردان 5/1 تا 5/2 برابر است. زنان نسبت به مردان به MS حساستر هستند به خصوص در طول 3 ماه اول پس از زایمان.
MS یک بیماری خود ایمنی بوده و در اصل سیستم اعصاب مرکزی شامل مغز و نخاع را درگیر میکند. خستگی، اختلال شناختی، افسردگی و خلق و خوی نامناسب بهعنوان علائم رایج بیماری MS شناخته شدهاند.
امروزه مطالعات نشان دادهاند که ضایعات زیاد و مشخصی در دستگاه ادراری بهعنوان علائم واضح در بیماران MS وجود دارد. از سوی دیگر، MS یک علت بسیار مهم ناتوانی با منشاء عصبی در بزرگسالان جوان است و افسردگی بیشترین اختلال روانشناختی مشاهده شده در MS میباشد. اختلالات شنوایی مرکزی و محیطی همیشه در MS دیده میشود. نوریت بینایی (التهاب، آماس یا زخم مربوط به عصب چشم) نیز معمولاً نشانهای از MS است. علائم MS وابسته به حمله عصبی خاص، در سیستم اعصاب مرکزی است و ممکن است در نهایت منجر به بیحسی و گزگز در عضلات، ضعف عضلانی، رفلکس ضعیف، اسپاسم عضلانی، مشکل در حرکت، عدم هماهنگی و عدمتعادل در برخورد با دیگران، مشکل در گفتار، مشکل بینایی، احساس خستگی، درد حاد یا مزمن و مشکلات مثانه و روده گردد. افسردگی همیشه همراه با MS و ناشی از خلق و خوی متغیر بیماران MS است. علاوه بر این، مشکلات تفکر واحساسی نیز در MS دیده میشود.
به طور کلی بیماری MS به چهار دوره طبقهبندی میگردد:
این نوع MS شایعترین نوع است که تقریبا ۸۵ درصد از افراد مبتلا به ام اس در زمان تشخیص به این نوع مبتلا هستند. در این نوع MS علائم قدیمی بدتر شده و ممکن است علائم جدید ظاهر شود. علايم خفیف تا شدید و همچنین عود و بهبودی روزها یا ماهها طول میکشد. این نوع ام اس خوشخیمتر از سایر انواع آن است و به نسبت، پاسخ دارویی بهتری میدهد و معمولا تا سن ۲۳ سالگی تشخیص داده میشود.
این نوع ام اس دوره عود و بهبود ندارد، به آرامی و به طور مداوم از زمان شروع آن پیشرفت میکند. علائم به یکباره اتفاق نمیافتد و به آهستگی پیش میرود. در این نوع ام اس علائم بدون کاهش شدت باقی میماند. تقریبا ۱۵ درصد از مبتلایان به ام اس به این نوع مبتلا هستند.
این نوع اماس فقط در مبتلایان به RRMS اتفاق میافتد. مرحله انتهایی ام اس عود کننده-فروکش کننده به صورت SPMS میباشد که حدود ۲۰ سال طول میکشد. در این نوع ام اس علائم بهطور مداوم از زمان شروع بیماری و به آرامی پیشرفت میکند، و بدون کاهش و بدون دوره بهبودی است. در این نوع ام اس به خود عصب حمله میشود.
این نوع ام اس بدخیمترین نوع ام اس است ولی شیوع بسیار اندکی دارد. در PRMS همانند نوع پیشروندهی اولیه، علائم همیشه در حال پیشرفت است و با عود و حمله نیز همراه است. در واقع این نوع ام اس ترکیبی از نوع عود کننده و پیشروندهی اولیه میباشد، با این تفاوت که همیشه پیشرفت میکند و با هر حمله وخیمتر میشود.
بررسی فاکتورهایی که باعث بروز حملات در افراد مبتلا به MS هستند و نیز فاکتورهایی که منجربه ایجاد MS میشوند بسیار مهم است. این فاکتورها به دو دسته تقسیم میشوند:
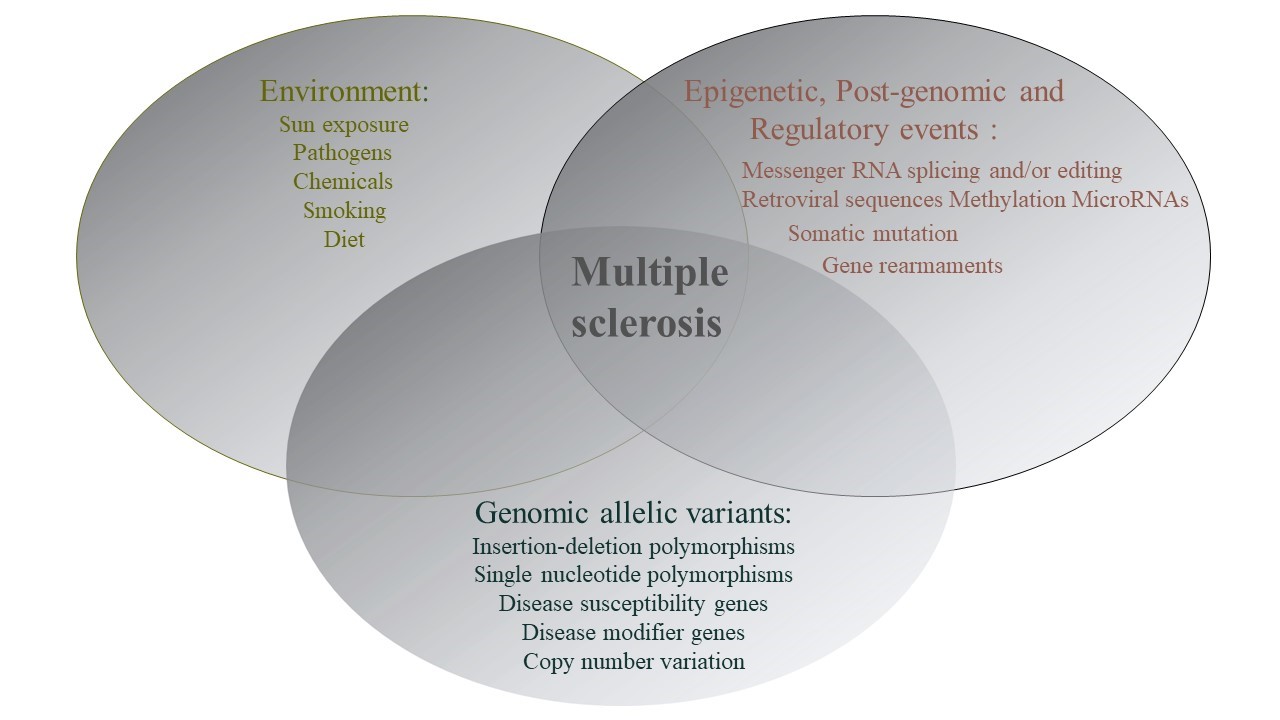
معمولا فاکتورهایی که باعث آغاز حملات MS میشوند ( نه همیشه ) فاکتورهای محیطی هستند نه ژنتیکی. چراکه ساختار ژنتیکی فرد در هر زمان ثابت بوده و عوامل محیطی میتوانند بر آنها تاثیرگذار باشند و مشخص کنند که آیا MS در فردی که از نظر ژنتیکی مستعد ابتلا به MS است بروز میکند یا خیر.
در ایجاد این بیماری، برهمکنش بین زمینه ژنتیکی فرد و شرایط محیطی که فرد در طول زندگیاش تجربه میکند، دخیل است
زمینهی ژنتیکی فرد مهمترین فاکتورخطر MS است. این بیماری تنها ناشی از یک ژن نیست و چندین ژن در ایجاد آن دخیل هستند. درحالیکه بزرگترین عامل ژنتیکی، مربوط به آنتیژن لوکوسیت انسانی کلاس دو استHLA class Ⅱ))، اما لوکوس دیگری مربوط به آنتیژن لوکوسیت انسانی کلاس یک نیز بهطور مستقل، تاثیرگذار است. درصد ابتلا به MS در فردی که عضوی از یک خانواده با سابقه MS است، به دلیل شباهت ژنتیکی، افزایش مییابد. این نسبت در بین خواهر و برادر که حدود 50 درصد شباهت ژنتیکی دارند، 20 تا 30 برابر نسبت به جمعیت عادی، افزایش دارد. دوقلوهای تک تخمکی که 100 درصد شباهت ژنتیکی دارند، نسبت به خواهر و برادر، تقریبا 10 برابر بیشتر است. تغییرات در ژنHLA-DRB1 قویترین عامل خطر ژنتیکی برای ابتلا به مولتیپل اسکلروزیس است و یکی دیگر از ژنهای مهم در بروز MS ژن IL-7R میباشد. هر دو ژن ذکر شده در سیستم ایمنی ایفای نقش میکنند و تغییرات در آنها ممکن است به پاسخهای خود ایمن منجر گردد و متعاقب آن، غلاف میلین و سلولهای عصبی دچار آسیب شوند و بیماری MS بروز پیدا کند. با این وجود هنوز مشخص نشدهاست که تغییرات درهر یک از این ژنها چه نقشی در ایجاد بیماری دارند. از دیگر ژنهایی که ممکن است در ایجاد MS دخیل باشند میتوان CYP27B1، TNFRSF1A، IL2RA و … نام برد.
بهطور کلی تغییرات ژنتیکی، یک شاخص مهم در استعداد ابتلا به مولتیپل اسکلروزیس(MS) و پیشرفت آن است.
MS جزء “بیماریهای ژنتیکی پیچیده ” محسوب میشود و در ایجاد این دسته از بیماریها برهمکنش ژنتیک و محیط ایفای نقش میکنند.
خوشهژن آنتیژن لوکوسیت انسانی، مهمترین لوکوس برای استعداد ابتلا به MS میباشد و این ژنها در مطالعات پیوستگی هم نشان داده شدهاند.
مطالعات GWAS فاکتورهای خطر متعددی را بهعنوان استعداد ابتلا به MS معرفی کردهاند، اما فقط تعداد کمی از آنها قابلیت توارث دارند.
با کمک فنآوریهای با ظرفیت بالا مانند NGS، میتوان مکانیسمهای دخیل در MS را تبیین کرد و به مدل بیماریزایی قابل اعتمادی میتوان دست پیدا کرد.
الگوی توارث مولتیپل اسکلروزیس مشخص نیست، اگرچه به نظر میرسد این وضعیت از طریق نسلها در خانوادهها منتقل میشود. خطر ابتلا به مولتیپل اسکلروزیس برای خواهر و برادر یا فرزندان یک فرد مبتلا به این بیماری بیشتر از جمعیت عمومی است.
هایپرپلازی مادرزادی آدرنال (CAH) یک اختلال ارثی غدد فوق کلیوی است که در هر دو جنس پسر و دختر ممکن است رخ دهد. غدد فوق کلیوی در بالای کلیهها قرار دارند و سه نوع هورمون تولید میکنند:
کورتیزول: کورتیزول یک استروئید است که توسط غدد فوق کلیوی تولید میشود و بدن ما برای مقابله با استرس فیزیکی و احساسی، تامین انرژی کافی و تنظیم میزان قند خون به آن نیاز دارد. غدد فوق کلیوی توسط غده هیپوفیز کنترل میشود.
آلدوسترون: حفظ حجم طبیعی مایعات بدن و تنظیم سدیم و پتاسیم بدن بر عهده این هورمون است. این یونها عملکرد متنوعی در بدن دارند و یکی از آنها تثبیت عملکرد قلب میباشد.
آندروژنها، گروهی از هورمونها هستند که تکوین ویژگیهای مردانه را کنترل میکنند.
افراد مبتلا به CAH توانایی تولید کورتیزول کافی را ندارند و در برخی موارد قادر به تولید آلدوسترون نیز نیستند. آنها همچنین مقدار زیادی آندروژن مانند تستوسترون و 17 هیدروکسی پروژسترون تولید میکنند. این عدم تعادل هورمونی میتواند منجر به بیماریهای جدی مانند اندام تناسلی غیر معمول، بلوغ زودرس، اختلال در رشد و سایر مشکلات شود.
در کودکان مبتلا به CAH، غدد فوق کلیوی فاقد آنزیمهای خاصی هستند که برای پردازش کورتیزول و آلدوسترون لازم است. در نزدیک به 95 درصد موارد، نقص در آنزیم 21 هیدروکسیلاز دیده میشود. CAH ناشی از نقص در این آنزیم، کمبود 21 هیدروکسیلاز نامیده میشود. دو شکل از هیپرپلازی مادرزادی آدرنال وجود دارد که ناشی از کمبود 21 هیدروکسیلاز است.
CAH کلاسیک: این شدیدترین شکل بیماری است و کمتر شایع است. در افراد مبتلا به CAH کلاسیک، بدن قادر به تولید کورتیزول نیست که برای تنظیم فشار خون، قند خون و کمک به بدن در واکنش به بیماری یا آسیب لازم است. بسیاری از کودکان مبتلا به CAH کلاسیک همچنین قادر به ترشح آلدوسترون یا حفظ مقدار کافی نمک در بدن خود نیستند، وضعیتی که در صورت عدم درمان میتواند منجر به کم آبی شدید و حتی مرگ شود.
CAH غیر کلاسیک: این رایجترین شکل CAH است و از نظر تظاهر خفیف است. بدن به اندازه کافی کورتیزول تولید میکند، اما مقدار زیادی آندروژن خاص مانند تستوسترون و 17 هیدروکسی پروژسترون تولید میکند. ترشح آلدوسترون در افراد مبتلا به CAH غیرکلاسیک طبیعی است.
| انواع CAH | ژن عامل بیماری | علائم بالینی |
|---|---|---|
| کمبود 21- هیدروکسیلاز | CYP21A2CYP21A2 & TNXB | کلاسیک: 46,XX دستگاه تناسلی مبهم، نارسایی آدرنال، هدر رفتن نمک، ویریل شدن (بروز صفات مردانه) پس از زایمان.غیرکلاسیک: هایپرآندروژنیسم (مقادیر بالای آندروژن) در دوران کودکی یا اوایل بزرگسالی ممکن است بدون علامت باشد.CAH-X: علاوه بر موارد فوق، حرکت بیش از حد مفاصل، درد مفاصل، دررفتگی مفاصل متعدد، نقایص خط میانی از جمله ناهنجاریهای ساختاری قلبی. |
| کمبود 21- هیدروکسیلاز | CYP11B1 | کلاسیک: 46,XX دستگاه تناسلی مبهم، ویریلیزاسیون پس از تولد، فشار خون بالا.غیر کلاسیک: هایپرآندروژنیسم در دوران کودکی یا اوایل بزرگسالی. ممکن است بدون علامت باشند. |
| کمبود 17-آلفا هیدروکسیلاز | CYP17A1 | کلاسیک: فنوتیپ زنانه (46,XX یا 46,XY وارونگی جنسی)، فشار خون بالا، تاخیر بلوغ با فقدان خصوصیات جنسی ثانویه.فرم جزیی (Partial) : 46,XY درجات متغیری از ابهام تناسلی. 46,XX بروز درجات متفاوتی از خصوصیات جنسی ثانویه. |
| کمبود3-بتا هیدروکسی دهیدروژناز تیپ 2 | HSD3B2 | کلاسیک: 46,XX و 46,XY ابهام دستگاه تناسلی، نارسایی آدرنال، هدر رفتن نمک. |
| کمبود اکسیدوردوکتاز P450 | POR | کلاسیک: 46,XX و 46,XY ابهام دستگاه تناسلی، نارسایی آدرنال، هدر رفتن نمک. |
| CAH لیپوئید | StAR | کلاسیک: فنوتیپ زنانه (46,XX یا 46,XY با وارونگی جنسی) نارسایی آدرنال، هدر رفتن شدید نمک.غیرکلاسیک: 46,XY با درجات متفاوتی از ابهام تناسلی، نارسایی آدرنال. |
| کمبود آنزیم برش زنجیره جانبی کلسترول | CYP11A1 | کلاسیک: فنوتیپ زنانه (46,XX یا 46,XY با وارونگی جنسی) ، نارسایی آدرنال، هدر رفتن نمک.غیرکلاسیک: 46,XY با درجات متفاوتی از ابهام تناسلی ، نارسایی آدرنال. |
کودکان مبتلا به CAH کلاسیک ممکن است دچار “بحران آدرنال” شوند که علائم زیر را ایجاد میکند:
استفراغ، کم آبی شدید، فشار خون پایین، شوک تهدید کنندهی زندگی در CAH کلاسیک. بدن همچنین آندروژن بیش از حد تولید میکند. افزایش تولید آندروژن میتواند باعث ابهام جنسی در دختران تازه متولد شده گردد. همچنین در کودکان بزرگتر باعث بلوغ زودرس، رشد سریع، قد کوتاه در بزرگسالی و مشکلات باروری میگردد.
کودکان مبتلا به CAH غیرکلاسیک ممکن است دارای علائم زیر باشند:
بلوغ زودرس، پوست چرب، آکنه، رشد سریع در دوران نوجوانی، قد کوتاه در بزرگسالی، پریودهای نامنظم در زنان و مشکلات باروری
هیپرپلازی مادرزادی آدرنال یک اختلال ژنتیکی است. در کودکان مبتلا به CAH، ژن 21-هیدروکسیلاز که آنزیم مورد نیاز برای تولید کورتیزول و آلدوسترون را میسازد، به درستی کار نمیکند. برای اینکه کودکی با CAH متولد شود، هر دو والدین باید ناقل ژن جهش یافته باشند و آن را به نوزاد خود منتقل کنند. CAH ناشی از کمبود 21-هیدروکسیلاز میتواند پسران و دختران را به طور یکسان تحت تاثیر قرار دهد. از هر 10000 تا 18000 کودک، یک کودک با CAH کلاسیک متولد میشود، در حالی که شکل غیر کلاسیک آن بسیار رایجتر است.
حذفها و جهشهای ژن CYP21A2 همه موارد شکل کمبود 21-هیدروکسیلاز CAH را تشکیل میدهند. جهش در ژنهای CYP11B1، CYP17A1، HSD3B2، CYP11A1، STAR و CYPOR باعث ایجاد فرمهای دیگر CAH میگردد.
تقریباً تمام اشکال CAH به صورت اتوزومال مغلوب به ارث میرسند. اختلالات ژنتیکی مغلوب زمانی رخ میدهد که یک فرد یک ژن غیرطبیعی را از هر یک از والدین به ارث میبرد. اگر فردی یک ژن طبیعی و یک ژن غیرطبیعی برای بیماری دریافت کند، فرد ناقل بیماری خواهد بود، اما معمولاً علائمی از خود نشان نخواهد داد. احتمال این که والدین ناقل هر دو ژن غیرطبیعی را منتقل کنند و در نتیجه فرزندی مبتلا داشته باشند در هر بارداری 25 درصد است. احتمال داشتن فرزند ناقل مانند والدین در هر بارداری 50 درصد است. شانس دریافت ژنهای طبیعی از هر دو والدین برای کودک 25 درصد است. این احتمالات برای مردان و زنان یکسان است.
شایع ترین شکل CAH، کمبود 21-هیدروکسیلاز، تقریباً 1:10000 تا 1:15000 نفر را در ایالات متحده و اروپا تحت تأثیر قرار میدهد. در میان اسکیموهای یوپیک، بروز شکل هدر دهنده نمک این اختلال ممکن است به 1 نفر از هر 282 نفر برسد. سایر اشکال CAH بسیار نادرتر هستند. در مقابل، CAH غیرکلاسیک تقریباً 1 در 100 تا 1 در 200 نفر در جمعیت عمومی را تحت تأثیر قرار میدهد.
همه نوزادان در ایالات متحده برای کمبود کلاسیک ۲۱-هیدروکسیلاز غربالگری میشوند. نوع غیر کلاسیک اغلب در آزمایش نوزاد تشخیص داده نمیشود و بنابراین، ممکن است تا دوران کودکی یا اوایل بزرگسالی که بیمار برای اولین بار علائم را نشان میدهد، تشخیص داده نشود. آزمایش ژنتیکی برای جهشهای ژنی مرتبط با اشکال مختلف CAH در دسترس است، اما اندیکاسیون آن زمانی است که در معاینات و آزمایشات متخصصین غدد ابهام هورمونی مشاهده شود و متعاقب آن زوجین توسط متخصص مشاوره ژنتیک بررسی شوند. تشخیص قبل از تولد برای زوجهایی که در معرض خطر ابتلا به CAH هستند، با استفاده از نمونهبرداری از پرزهای جفتی سه ماهه اول بارداری و آزمایش DNA جنین برای جهش ژنی خاص CAH که در خانواده رخ میدهد، در دسترس است. تعیین جنسیت غیر تهاجمی را میتوان از طریق آزمایش DNA جنین در خون مادر انجام داد. آزمایش غیر تهاجمی برای بررسی وضعیت جهش در ژنهای مرتبط با CAH فعلاً وجود ندارد.
اگر CAH در جنین تشخیص داده شود، درمان پیش از تولد امکان پذیر است. داروی خوراکی دگزامتازون را میتوان به زنان باردار در بارداریهای بعدی در صورتی که فرزندی با CAH کلاسیک شدید بدنیا آورده باشد تجویز کرد. چنین درمانی از بیماری پیشگیری یا آن را درمان نمیکند، اما ممکن است باعث کاهش ویریل شدن (virilization) جنینهای دختر مبتلا شود. پایش سطح هورمونها در طول زندگی از اهمیت ویژهای برخوردار است. ترشح بیش از حد هورمونهای جنسی میتواند باعث پیری زودرس استخوانها شود و در صورت تشخیص زودهنگام میتوان سرعت آن را کاهش داد.
تستهای مبتنی بر PCR با یا بدون توالییابی و MLPA بعنوان استاندارد طلایی برای تشخیص موتاسیونهای ژن CYP 21A2 شناخته میشوند. از سایر تکنیکهایی که برای تشخیص این بیماری استفاده شدهاند میتوان انواع مختلف PCR، سادرن بلاتینگ و NGS را نام برد. از این تکنیکها میتوان هم در تشخیص پیش از تولد (PND) و هم در تشخیص پیش از انتقال جنین به رحم (PGD) استفاده کرد. تکنیک PGD که همراه با IVF انجام میشود یک راهکار مناسب برای پیشگیری از انتقال ژنهای معیوب به نسل بعد میباشد. در تشخیصهای قبل از تولد تشخیص زود هنگام ژن SRY در خون مادر قابل انجام میباشد وامکان تعین جنسیت جنین در مراحل اولیه بارداری فراهم میشود. مشخص شدن جنسیت جنین باعث میشود صرفاً جنینهای دختر تحت درمان قرار گیرند(برای پیشگیری از ابهام تناسلی).
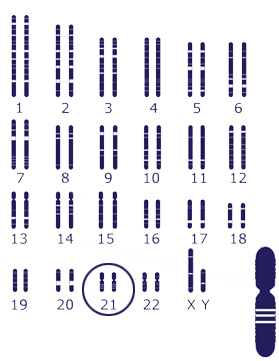
حذف، اضافه شدن یا بی نظمی درDNA موجود در ساختمان کروموزوم را ناهنجاری کروموزومی میگویند و زمانی اتفاق میافتد که هنگام میوز یا میتوز خطایی در تقسیم سلولی ایجاد شود. این ناهنجاریها را میتوان به انواع تعدادی، ساختاری وترکیبهای کروموزومی متفاوت در دو یا چند ردهی سلولی دستهبندی کرد.
کم یا اضافه شدن یک یا چند کروموزوم است وشامل آنیوپلوئیدی ( مونوزومی، تریزومی، تترازومی و …) و یوپلوئیدی (تریپلوئیدی، تتراپلوئیدی و…) میباشد.
ریزومیها، مانند سندرم داون (تریزومی 21)، سندرم پاتو (تریزومی 13)، سندرم ادوارد (تریزومی 18) ، سندرم کلاین فلترXXY)،47 ) سندرم جاکوب,XYY) 47) و سندرمXXX .
اکثر موارد تریزومیها در اوایل حاملگی سقط میشوند، مانند تریزومی 16 که یک یافتهی معمول درسقط های خودبخودی در سه ماههی اول میباشد. اضافه شدن یک کروموزوم جنسی تنها اثرات فنوتیپی خفیفی به همراه دارد.
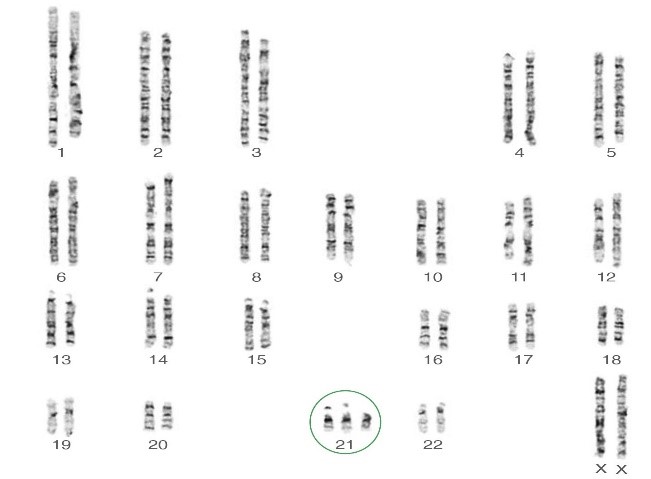
مونوزومی برای یک کروموزوم غیر جنسی تقریبا با بقا تا زمان تولد ناسازگار است. سندرم ترنر45,X مثالی برای مونوزومی کروموزومهای جنسی است .
سلولهای پلیپلوئیدی، شامل چندین مجموعه هاپلوئیدی از کروموزومها میباشند. مانند تتراپلوئیدی ( 92 کروموزوم) و یا تریپلوئیدی (69 کروموزوم) که در انسان اغلب در موارد سقط خودبخودی دیده میشود.

نوآرایی ساختار کروموزوم که حاصل شکستگی کروموزوم واتصال مجدد آن با آرایش متفاوت است، که میتواند متعادل یا نا متعادل باشد. مانند جابجاییها، معکوس شدگی، اضافه شدنها، حذفها، کروموزومهای حلقوی و ایزوکروموزومها.
ابجایی به انتقال مادهی ژنتیکی از یک کروموزوم به کروموزوم دیگر گفته میشود. مانند روبرت سونین.

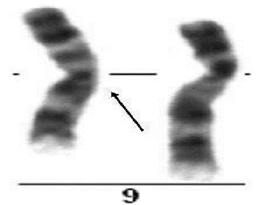
عکوس شدگی یا واژگونی، زمانی اتفاق میافتد که قطعهای از کروموزوم، وارونه شود. به عنوان مثال واژگونی پریسانتریک در کروموزوم 9 که یک پلیمورفیسم نرمال است.
اضافه شدن هنگامی اتفاق میافتد که قطعهای از کروموزوم درون کروموزوم دیگری وارد شود.
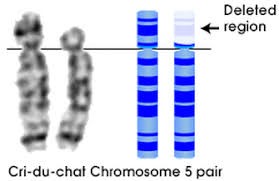
ذف شامل فقدان قسمتی از کروموزوم است. مانند سندرم ولف هیرشهورن و سندرم فریاد گربه.
روموزومهای حلقوی زمانی تشکیل میشوند که، یک شکستگی در هر کدام از بازوهای یک کروموزوم رخ دهد و دو انتهای چسبندهی آن به صورت حلقه به هم متصل شوند.
ایزوکروموزم زمانی ایجاد میشود که یک بازوی کروموزوم حذف و بازوی دیگر مضاعف شود. شایعترین ایزوکروموزوم مشاهده شده، ایزوکروموزوم X با دو بازوی بلند است.

به حضور دو یا چند ردهی سلولی با ساختار ژنتیکی متفاوت در یک فرد گفته میشود که این سلولها از یک سلول تخم منفرد ایجاد شدهاند.
زمانی که دو یا چند ردهی سلولی با ساختار ژنتیکی متفاوت در فردی دیده شود که این سلولها از بیش از یک سلول منشا گرفته باشند، کایمریسم ایجاد میشود.
سیتوژنتیک علم مطالعه ساختمان کروموزومهاست. در این علم کروموزومها با استفاده از تکنیکهای باندینگ (نوارگذاری) و یا شیوههای سیتوژنتیک مولکولی، مورد تحلیل و بررسی قرار میگیرند.
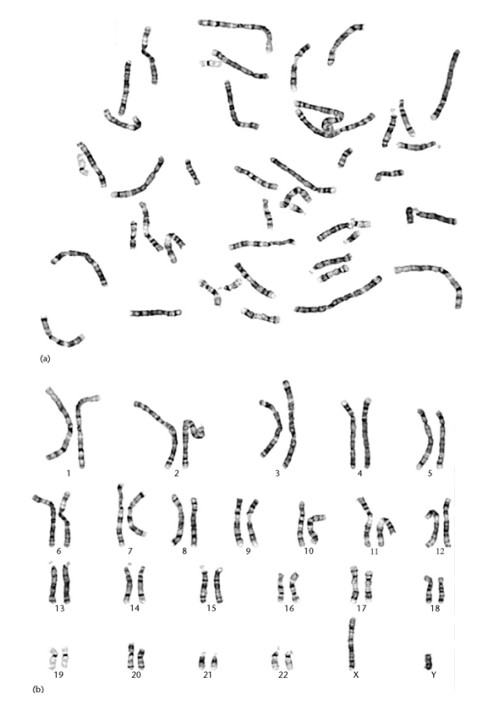
این آزمایش به بررسی تغییرات کروموزومی پرداخته و در واقع تصویری از تعداد و ساختار کروموزومهای فرد ارائه میدهد، که میتوان به وسیلهی این بررسی به بسیاری ازاختلالات و ناهنجاریهای کروموزومی پی برد. آزمایش کاریوتایپ میتواند روی نمونه خون، مغز استخوان، بافت و یا نمونه جنینی انجام گیرد. آزمایش کاریوتایپ روی نمونههای خون به صورت معمولی و با تفکیکپذیری بالا، انجام میگیرد. مراحل انجام آزمایش شامل کشت، هاروست، لامگیری یا تهیهی گستره کروموزومی، رنگآمیزی کروموزومها و بررسی دقیق زیر میکروسکوپ میباشد. برای کشت کروموزومی خون محیطی، باید تا حد امکان از خون تازه که به آن ماده ضد انعقاد سدیم هپارین اضافه شده است استفاده شود. کشت کروموزومی معمولاً ۷۲ ساعت وقت نیاز دارد.
سیتوژنتیک مولکولی شامل روشهای زیر است:
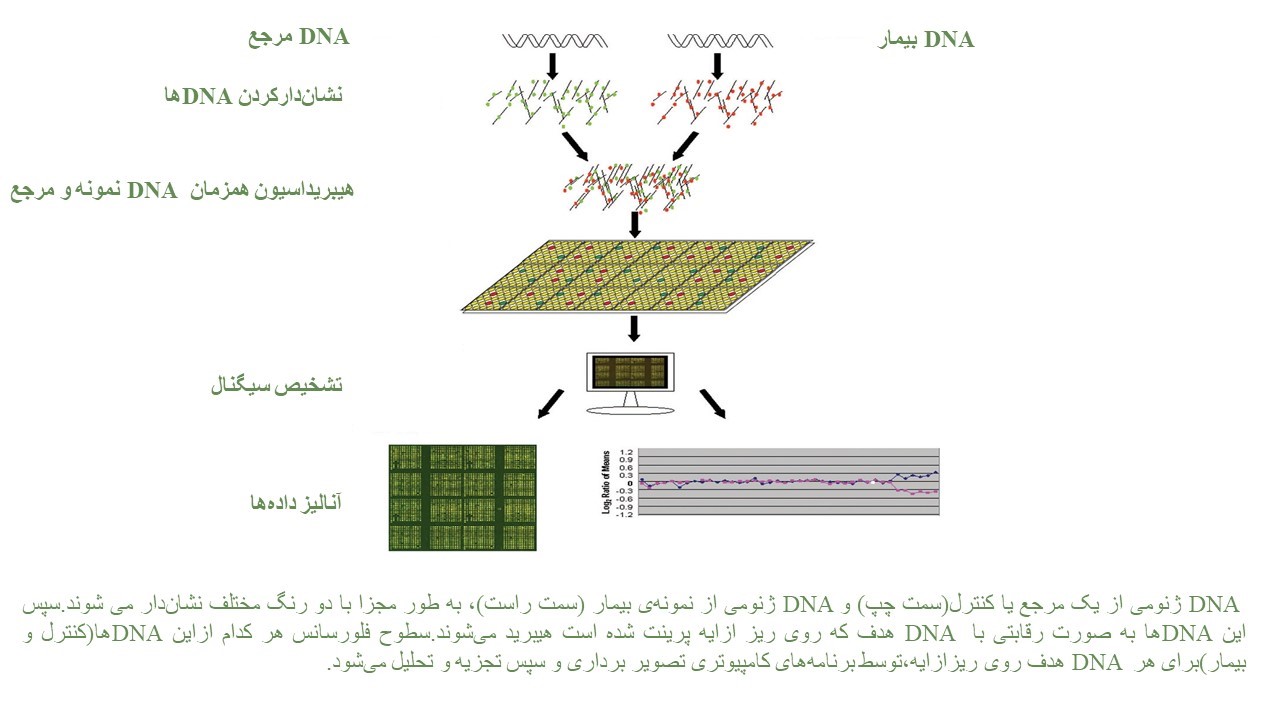
CMA شامل دو روش array CGH وSNP array میباشد و جهت شناسایی عدم تعادل ژنتیکی در نوزادان و کودکان با GDD (Global Developmental Delay) ،اُتیسم، ناهنجاریهای مادرزادی و… ارزش تشخیصی بیشتری دارد. انجام این آزمایش روی بافت جنینی جفت برای بررسی علت سقط در اوایل بارداری، سقط مکرر، مرگ جنین داخل رحم مادر نیز کاربرد دارد.
تکنيکي با قدرت تفکيک بالا، براي بررسي حذفها و اضافه شدگيها در کل ژنوم و شناسايي عدم تعادل کروموزومی است. اين آزمايش را کاريوتيپ مولکولي مينامند و به ويژه براي شناسايي علت عقب ماندگیهای ذهنی و يا ناهنجاریهای مادرزادی کاربرد دارد.
با استفاده از این روش میتوان حذف یا اضافهشدن تعداد نسخههای ژنومی را با دقت بالا تشخیص داد. انجام SNP در افراد مبتلا به ناهنجاریهای مادرزادی متعدد، تاخیر رشد، ناتوانی ذهنی و اوتیسم توصیه میشود. علاوه بر این موارد،SNP میتواند مناطق خنثی هموزیگوسیتی (ROHs) را شناسایی کند، که میتواند حاکی از افزایش خطر اختلالات اتوزومال مغلوب به دلیل وجود ژنهای واقع در ROH، دیزومی تکوالدی یا خویشاوندی میباشد.
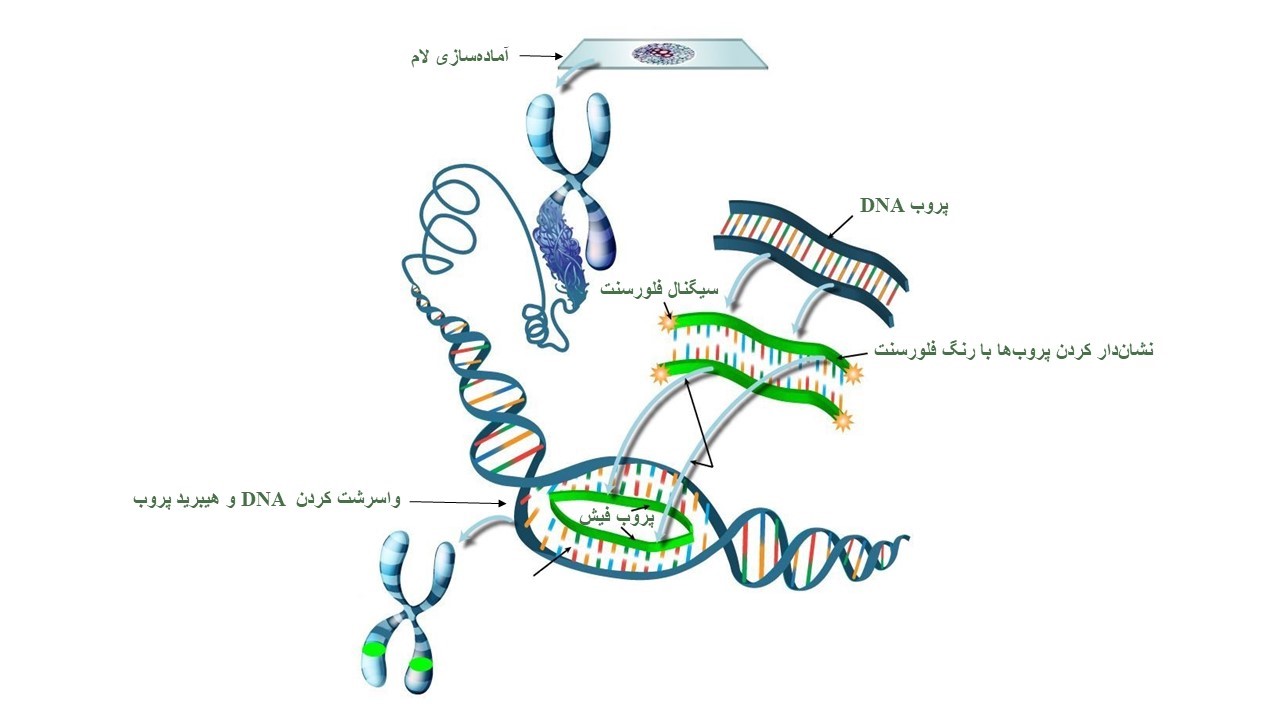
این روش یک تکنیک سیتوژنتیکی است که از سال 1980 ارائه شده است. در روش FISH قسمتهای خاصی از کروموزومها با استفاده از پروبهایی که با مادهی فلورسنت نشاندار شدهاند، رنگآمیزی میشوند و از این طریق میتوان به ناهنجاری موجود در آن ناحیه پی برد. پروبهای مورد استفاده در این روش قطعهای از DNA یاRNA هستند که مکمل توالیهای ویژهای برروی کروموزومها میباشند و به آنها متصل میشوند. با استفاده از این روش ناهنجاریهای کروموزومی که با روشهای سیتوژنتیک معمولی قابل تشخیص نیست را میتوان بررسی کرد. در ابتدا مجموعهی کامل کروموزومهای یک فرد روی یک اسلاید شیشهای تثبیت میشود و سپس در معرض قطعات کوچک پروب قرار میگیرد. این پروبها توالی همسان خود را در مجموعهی کرموزومها پیدا کرده و به آن متصل میشوند و توسط میکرسکوپ مخصوصی مطالعه میشوند.
این تکنیک، یک روش جدید و پیشرفته است که میتواند تغییر در تعداد کپیهای چندین ژن را بصورت همزمان به اثبات رساند. MLPA در تشخیص مولکولی بیماریهای ژنتیکیای که به علت حذف یا مضاعفشدگی ژنهای خاص ایجاد میشود کاربرد دارد. این حذف یا مضاعفشدگیها ممکن است اثر فنوتیپی کاملاً متفاوتی ایجاد کنند. برای مثال در بیماری دیستروفی عضلانی دوشن( DMD ) ، حذف و مضاعفشدگی در برخی اگزونها اتفاق میافتد نه در کل ژن. علاوه بر این فقدان کامل پروتئین منجر به DMD و وجود پروتئین جزئی باعث BMD میشود. روش MLPA قادر است تا 55 توالی هدف را در یک واکنش PCR تجزیه و تحلیل کند. هر کدام از این توالیها پروب متفاوتی نیاز دارند که خود از دو پروب در جهتهای معکوس تشکیل شدهاست.(LPO, RPO)
یکی از مزیتهای این روش این است که به مقدار ناچیزی از DNA برای تشخیص نیاز است (حدود 50 نانوگرم).
علاوه بر این، MLPA میتواند درتشخیص مولکولی بیماریهای ژنتیکیای که به دلیل متیلاسیون غیر طبیعی DNAبوجود میآید استفاده شود. (MS-MLPA) این روش نیازی به تبدیل بی سولفیت سدیم باقیماندههای سیتوزین متیله شده ندارد و در عوض از اندونوکلئاز HhaⅠ استفاده میکند. اگر محل تشخیص HhaⅠ متیله نباشد، این آنزیم هیبرید DNA پروب- نمونه را قطع میکند و محصول PCR تشکیل نمیشود.
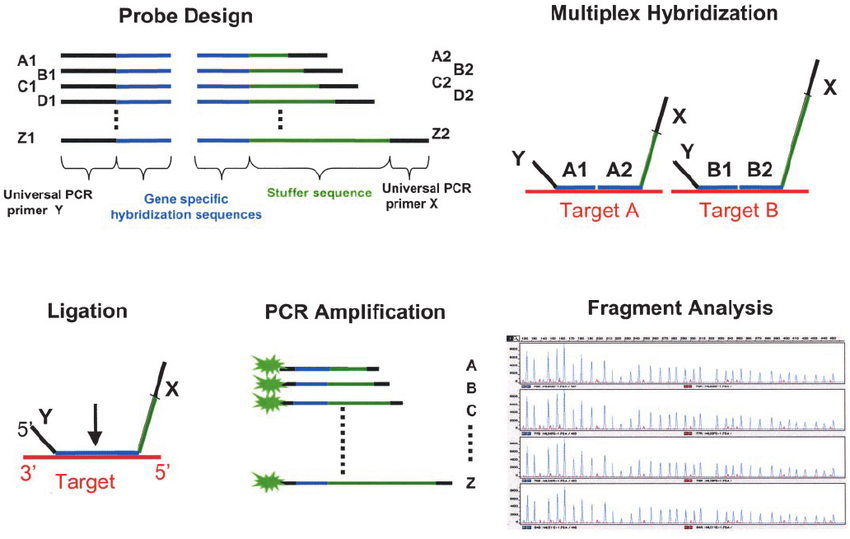
مطالعهی کروموزومها و تقسم سلولی، سیتوژنتیک نامیده میشود. هر بافت با سلولهای زنده و هستهدار قادر به تقسیم میتواند برای مطالعهی کروموزومهای انسان به کار رود. اگرچه نمونهی مناسب آنالیز کروموزومی را میتوان به سهولت از پوست، مغزاستخوان، پرزهای کوریونی یا سلولهای مایع آمنیوتیک (آمنیوسیتها) نیز به دست آورد، ولی معمولترین آنها لنفوسیتهای خون محیطی است. انتخاب بافت بسته به بیمار متفاوت است، بهعنوان مثال برای تشخیص پیش از تولد در جنین، از مایع آمنیوتیک(Amniotic Fluid) و پرزهای جفتی(Chorionic Villus) استفاده میگردد. برای بالغین و نوزادان از خون محیطی (Peripheral Blood)، و به میزان کمتری از مغز استخوان، پوست وبافتهای دیگر نیز استفاده میشود. در مواردی که ناهنجاریهای کروموزومی، اکتسابی و با Neoplasms در ارتباط باشد، از بافتی که در بدخیمی درگیر است برای بررسی میتوان استفاده کرد. تمام بافتهایی که برای تهیه کروموزوم به کار میروند در محیط کشت در آزمایشگاه، کشت داده میشوند. آزمایش کاریوتایپ برای توضیح فتومیکروگراف کروموزومهای یک فرد که به روش استاندارد مرتب شده انجام میشود. این آزمایش به بررسی تغییرات کروموزومی پرداخته و در واقع تصویری از تعداد و ساختار کروموزومهای فرد ارائه میدهد که به وسیلهی آن بسیاری از اختلالات و ناهنجاریهای کروموزومی تشخیص داده میشود. مراحل انجام آزمایش شامل کشت، هاروست، لامگیری یا تهیهی گسترهی کروموزومی، رنگ آمیزی کروموزومها و بررسی دقیق زیر میکروسکوپ میباشد.
تجهیزات و فضاهای لازم در آزمایشگاه سیتوژنتیک:
هر آزمایشگاه سیتوژنتیک، لازم است که یک محل کشت سلول جداگانه و استریل داشته باشد. بنابراین بیشتر آزمایشگاهها از هودهای با جریان Laminar استفاده میکنند. در هودهای Laminar میبایستی یک لامپ UV و جریان هوای فیلتر شده تعبیه شده باشد. این هود باید در جایی قرار گیرد که حداقل رفت و آمد و شلوغی را داشته باشند.
یک هود شیمیایی جهت تهیه و کاربا محلولها
بهتر است فضایی محفوظ جهت کنترل دما و رطوبت، در نظر گرفته شود.
دستگاه انکوباتور، فور، یخچال و فریزر، دستگاه سانتریفوژ، PH متر، دستگاه بخور گرم، میکروسکوپ نوری و اینورت، ، رطوبتسنج و دماسنج، بن ماری و سینک.
نمونه خون محیطی را به حجم کمی از محیط کشت حاوی سرم جنین گاوی(FBS) و فیتوهماگلوتینین (PHA) اضافه میکنند که لنفوسیتهای T را تحریک به تقسیم مینماید. برای کشت کروموزومی خون محیطی، باید در حد امکان از خون تازه که به آن ماده ضد انعقاد سدیم هپارین اضافه شده است، استفاده شود. کشت کروموزومی معمولا ۷۲ ساعت زمان نیاز دارد.
سلولها تحت شرایط استریل در دمای C˚٣٧ و به مدت ٣ روز کشت داده میشوند و سپس کلسمید به محیط کشت اضافه میشود. این ماده ویژگی بسیار سودمندی در جلوگیری از تشکیل دوک داشته، بهطوری که تقسیم سلولی را طی متافاز، زمانی که کروموزومها به حداکثر فشردگی خود رسیدهاند و بنابراین قابل مشاهدهاند، متوقف میکند. در ادامه نمک هیپوتونیک اضافه میشود که باعث لیز سلولها میشود. سپس با مخلوط متانول و اسیداستیک (فیکس) شستشو انجام گرفته و لامگیری صورت میگیرد. در مرحلهی بعد که Aging نامیده میشود لامها به مدت چند ساعت تا چند روز در دمای مشخصی قرار داده میشوند. اینکار جهت بالا بردن وضوح بندها است. مرحلهی بعد Banding است. یک بند به این صورت تعریف میگردد: قسمتی از کروموزوم که از طریق تیرهتر شدن یا روشنتر شدن توسط یک یا چند تکنیک بندینگ، به وضوح از قطعات مجاورش قابل افتراق میباشد. باندهایی که به وسیلهی یک روش خاصی رنگ تیره به خود میگیرند، ممکن است با دیگر روشهای رنگآمیزی رنگ روشن پیدا نمایند. در روشهای بندینگ اولیه، به منظور تولید یک الگوی باندی فلورسنت، از کیناکرین موستارد یا دیهیدروکلرید کیناکرین استفاده میگردید. این روش بر پایهی الگوی بندینگ Q میباشد. روش دیگر بندینگG-bands نام دارد، که درآن معمولا از مخلوط رنگ گیمسا به عنوان مادهی رنگآمیزی استفاده میشود.
در برخی از تکنیکهای بندینگ، الگوهایی ایجاد میشود که شدت رنگگیری آنها، متضاد با الگوهای ایجاد شده با روش G-bands است، این روشها، روشهای معکوس R-bands نامیده میشوند.
(1) دسته اول باندهایی را ایجاد میکنند که در طول یک کروموزوم کامل وسالم، انتشار یافتهاند و شامل تکنیکهایی میباشند که الگوهای همانندسازی DNA را نشان میدهند که از جمله آنها میتوان R-bands ، Q-bandsوG-bands را نام برد.
(2) دسته دوم اجزا ساختمانی خاصی از کروموزومها را رنگآمیزی میکنند و لذا منجر به ایجاد تعداد محدودی از باندها میشوند. این روشها شامل روشهایی میباشند که هتروکروماتین اجباری (C-bands)، باندهای تلومریک (T-bands) و نواحی ساماندهی نوکلئولوس(NORs) را آشکار میسازند.
روش رنگآمیزی با گیمسا معمولترین آنهاست. در این روش کروموزومها ابتدا با تریپسین تیمار میشوند تا محتوای پروتئینی آنها دناتوره شود . هر باند در این روش تقریبا حاوی ۵ الی ١۰ میلیون باز است و ممکن است در برگیرندهی صدها ژن باشد. رنگ گیمسا از ائوزین آنیونی و تیازین کاتیونی (1:2) تشکیل شده است. رسوب رنگ در کروموزومها در مناطقی تشکیل میشود که فسفاتهایDNA در فاصلهی صحیح از هم قرار گرفته باشند، تا دو مولکول تیازین را به هم متصل کنند و سپس به مولکول ائوزین متصل میشوند. به طور کلی باندهای مثبت گیمسا (نواحی تیره) که غنی از AT هستند، کمتر رونویسی شده، تراکم بیشتر و ژنهای اندکی دارند. این مناطق آبگریز هستند. پروتئینهای آبگریز نواحی متراکم را حفظ میکنند و رسوب ترکیب تیازین–ائوزین را تسهیل میکنند. باندهای منفی گیمسا (نواحی روشن) که غنی از GC هستند، بیشتر رونویسی میشوند، تراکم کمتر و ژنهای بیشتری دارند. این مناطق آبگریزی کمتری داشته و در نتیجه کمتر برای رسوب تیازین-ائوزین مناسبند. درجهی وضوح به وسیلهی تعداد باندهای مشاهده شده در یک مجموعهی هاپلویید معین میگردد.
مرحلهی بعدی مهمترین مرحله و قسمت تشخیصی آزمایش کاریوتایپ است که شامل انتخاب گسترهی متافازی، شمارش تعداد کروموزومها و سپس آنالیز دقیق الگوی نواربندی کروموزومها است.
برای اکثر لوسمیها، به جز برخی موارد که از خون محیطی استفاده میشود، از نمونه مغز استخوان استفاده میکنند. سلولهای مغزاستخوان با رشد سریع، باید در معرض مواد غذائی بیشتری قرار بگیرند.
کشت مغز استخوان به دو صورت کشت معمولی و کشت قدرت تفکیک بالا صورت میپذیرد.کشت معمول خود به سه صورت زیر انجام میپذیرد.
کشت مستقیم (direct): به لولهی حاوی محیط کشت کامل و نمونه کلسمید اضافه شده و بعد از 30-60 دقیقه هاروست می شود.
کشت کوتاه مدت (24-48-72 ساعته): بعد از مدت زمان مورد نظر به لولهها کلسمید اضافه و بعد از 30 دقیقه هاروست میشود.
کشت کلسمید در طول شب(ONC): حدود 20 میکرولیتر کلسمید در ساعت 5 یا 6 عصر به لولهی حاوی محیط کشت و نمونه اضافه میشود و صبح روز بعد هاروست میگردد.
برای به دست آوردن کروموزومهای بلندتر باید سیکل سلولی را در مرحلهی S بوسیلهی یک مهار کننده مانند تیمیدین متوقف کرد.بعد از حدود 15 ساعتمهار کننده از طریق شستشو خارج شده و سپس بعد از حدور 4 الی هفت ساعت کلسمسد اضافه میشود و بعد از گذشت حدود 20 دقیقه هاروست انجام میگردد.
انتخاب نوع کشت بر اساس علت مراجعه صورت میپذیرد.
مایع آمنیوتیک در هفته 17 -15 حاملگی، توسط پزشک متخصص زنان به میزان 15-20 سیسی گرفته و در دو لوله پلاستیکی استریل قابل سانتریفوژ تقسیم میشود. نمونهها به سرعت و در دمای مناسب به آزمایشگاه منتقل میشود. مایع آمنیوتیک به رنگ زرد کمرنگ میباشد و اغلب در مراحل بعدی حاملگی وقتی غلظت سلولها بالاتر است، رنگ مایع کدر میشود. ابتدا لولههای حاوی نمونه سانتریفوژ شده و محلول رویی برای آنالیز تستهای شیمیایی فریزمیشود. رسوب باقی مانده را با تکان آرام دوباره به حالت محلول در اورده وسه سیسی محیط مناسب برای هر کشت اضافه میشود .بهتر از محیط کشتهای مختلف برای هر کشت استفاده کرد. دو فلاسک کشت داده میشود. کشتها به مدت 7-6 روز انکوبه میشوند. سپس رشد سلولها با استفاده از یک میکروسکوپ invert بررسی میگردد. در صورت مشاهدهی کلونی محیط برداشته شده و با محیط تازه تعویض میگردد.هر دو روز یکبار نمونهها با میکروسکوپ بررسی وو محیط آن تعویض میشود.بعد از رشد کلونی به اندازهی کافی، می توان یکی از لولهها را هاروست و لولهی دیگر را subculture کرد.به این صورت که محیط در لولهی جداگانهای ریخته میشود سپس به نمونه یک سیسی ازمحلول حاوی EDTAو تریپسین اضافه میشود.در این مرحله برای جدا شدن سلولها ضربه زده شود.سپس دو بار باPBS شستشو انجام شده و مجددا محلول trypsin-EDTA اضافه شده و بعد از ضربه محیط کشت اضافه میگردد. با جدا شدن سلولها از سطح ظرف کشت،برای خنثی کردن اثر تریپسین یک سیسی محیط کامل حاوی سرم اضافه میشود در این مرحله با توجه به مقدار نمونه، درون ظرفهای دیگر آن را aliquot کرده و 3 سیسی محیط تازه اضافه میشود و انکوبه میگردد.
جهت هاروست به ازای هر سیسی نمونه حدود 75میکرولیتر کلسمید اضافه میگردد وپس از حدود سه ساعت انکوبه کردن مرحلهی تریپسینه کردن انجام می شود و سوسپانسیون به دست امده15 دقیقه با دور 1000rpm سانتریفوژ و سپس هاروست میشود.
فانکونی: فانکونی نوعی کمخونی است که با علائم بالینی متنوعی همراه میباشد. نحوهی توراث این بیماری اتوزومال مغلوب است. سلولهای بیماران FA نسبت به عوامل پیوند متقابلDNP، مانند مایتومایسینC (MMC) و دی اپوکسی بوتان (DEB) یا سیس پلاتین، حساس هستند و در مواجهه با دارو به صورت مهار بیش از حد، توقف چرخهی سلولی و شکستگی کروموزومی، آشکار میشوند.
در این آزمایش، لنفوسیتهای Tموجود در نمونه در حضور مایتومایسین کشت داده میشوند. بهتر است برای هر بیمار، از یک کنترل منفی، ترجیحاً همسن و جنس مشابه استفاده شود. چهار کشت برای بیمار و کنترل منفی انجام میشود. یک کشت HR بدون مایتومایسین و سه کشت با غلظتهای متفاوت از مایتومایسین گذاشته میشود. سپس نمونهها هاروست و رنگآمیزی میشوند. پس از آن شکستها و ناهنجاریها در زیر میکروسکوپ بررسی میگردد. اگر تعداد شکستهای کروموزومی نسبت به نمونهی کنترل ۱/۰ بیشتر باشد تشخیص کمخونی فانکونی مثبت است.

سندرم ایکس شکننده یا مارتین بل، که بیشتر به عنوان یک بیماری تکژنی شناخته میشود، یک بیماری ارثی است که از والدین به فرزندان منتقل میشود. این بیماری یکی از شایعترین علل عقبماندگیهای ذهنی میباشد. این بیماری در پسران بیشتر مشاهده شده و علائم شدیدتری دارد.
این بیماری به این دلیل سندرم ایکس شکننده نامیده میشود، که در گسترهی کروموزومی این بیماران در انتهای بازوی بلند کروموزوم ایکس یک مکان شکننده دیده میشود. تقریبا در تمام موارد این بیماری جهش در بالادست ژن FMR1 و به دنبال آن بسط تکرارهای سه نوکلئوتیدی CGG در درون این ژن و متیلاسیون غیرطبیعی آن رخ میدهد. دفعات این تکرار در افراد ممکن است متفاوت باشد. در افراد طبیعی تعداد این تکرارها ده تا پنجاه بار است در حالیکه در افراد مبتلا تعداد این تکرارها ممکن است به بیش از دویست بار برسد. افزایش غیرطبیعی این تکرارها منجر به خاموشی ژن FMR1 شده و در نتیجه پروتئین FMRP تولید نمیگردد. شدت اختلال با تعداد این تکرارها نسبت مستقیم دارد.
یک سوم از بیماران مبتلا به سندرم ایکس شکننده ویژگیهای بیماری اتیسم را نیز نشان میدهند.
برای بررسی سندروم X شکننده سه نوع کشت صورت میگیرد: برای انجام این آزمایش سه کشت انجام میشود. کشت اول بصورتHR (72 ساعته)، کشت دوم با محیط کشت Low folate همراه با سرم گاوی، و کشت سوم به همراه تایمیدین اضافه (92 ساعته). ادامه مراحل آزمایش همانند بالا انجام و در نهایت لامهای آماده شده در زیر میکروسکوپ بررسی و مطالعه میشوند.
تشخیصهای مولکولی در سالهای اخیر وارد حوزههای درمانی شدهاند و با تغییر در نگرش و بینش متخصصان در مورد بیماریها، باعث ایجاد تحول در مراقبتهای بهداشتی گردیدهاند. درآزمایشگاههای تشخیصی مولکولی، مواد ژنتیکی یا پروتئینهای مرتبط با یک بیماری یا یک شرایط خاص مورد شناسایی و ارزیابی کمی و کیفی قرار گرفته میشوند. در این شرایط درک مکانیسمهای اساسی بیماریها، تسهیل شده و برای پزشکان برای درمانهای کارآمدتر راهگشا خواهد بود. پیشرفت مستمر در تکنولوژی، سرعت و عملکرد تشخیص مولکولی را افزایش داده و امروزه تعیین توالی کل ژنوم تقریباً برای همگان در دسترس میباشد. قابلیت انجام تستهای مولکولی بصورت اتوماتیک این امکان را فراهم آورده که این نوع از تستها برای کل مناطق جهان در دسترس باشند. تشخیص مولکولی بخش گسترده از تستهای تشخیصی را در برمی گیرد که به معنای واقعی کلمه سلامت فرد در سطح مولکولی ارزیابی میشود. در این تکنیکها توالیهای خاصی در DNA، RNA و پروتئینها شناسایی و اندازه گیری میشوند. این تکنیکها همچنین میتوانند حضور ویروسها ، باکتریها و سایر سلولهای خاص را شناسایی و اندازه گیری نمایند.
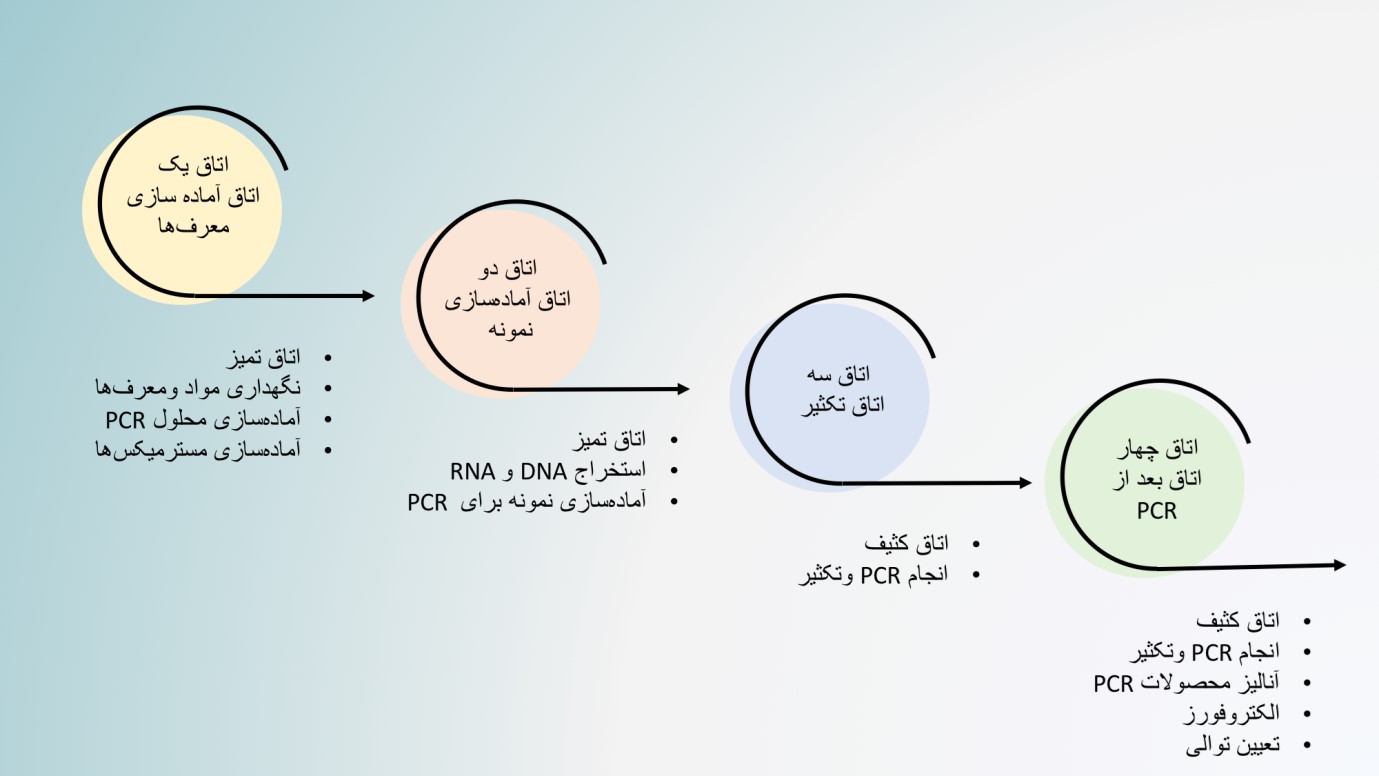
تاسیس آزمایشگاه مولکولی به فضای کافی، تجهیزات فنی مناسب و نیرو انسانی واجد شرایط نیاز دارد. الزامات تاسیس یک آزمایشگاه استاندارد با توجه به نوع فعالیت آزمایشگاه میتواند متفاوت باشد ولی چندین معیار اساسی وجود دارد که بهتر است رعایت شوند. یکی از مهمترین نکاتی که در طراحی آزمایشگاه مولکولی باید مورد توجه قرار گیرد، شرایط فیزیکی آزمایشگاه است. روشهای مبتنی بر PCR به آلودگی بسیار حساس هستند و هدف اصلی در طراحی فضاهای آزمایشگاه مولکولی جلوگیری از ایجاد آلودگی است. به دستآوردن مقدار زیادی محصول PCR از یک الگوی بسیار کم یک مزیت ویژه برای PCR محسوب میشود اما همین ویژگی در صورت وجود آلودگی منجر به نتایج نادرست و کاذب میگردد. نتایج مثبت کاذب ممکن است ناشی از انتقال از یک نمونه به نمونه دیگر، آلودگی متقابل از نمونههایی که بطور همزمان آماده میشوند ، محصولات PCR قبلی و آلوده بودن محلولها و مواد مورد استفاده باشد. در روش Real Time PCR معمولاً لولهها بسته میمانند و خطر ایجاد آلودگی کاهش مییابد. با توجه به اینکه تکنیکهای اصلی مورد استفاده در آزمایشگاههای مولکولی مبتنی بر PCR هستند، میتوان آنها را به دو فرآیند اصلی قبل از PCR(آماده سازی نمونه و آماده سازی مخلوط PCR) و بعد از PCR(انجام PCR و آنالیزهای بعد از آن) تقسیمبندی نمود. ضروری است این مراحل در فضاهای جداگانهای تحت عنوان “تمیز” و “کثیف” انجام شوند. ناحیهی تمیز در واقع ناحیهای است که تمام مراحل قبل از PCR مانند پردازش نمونه، استخراج DNA/RNA و آماده سازی PCR در آن انجام میشود. ناحیه کثیف هم فضایی است که در آن محصولات PCR(Amplicons) تولید و مورد ارزیابی قرار گرفته میشوند. کارکنان آزمایشگاه باید تمام معرفها ، مواد و تجهیزات موجود در این فضاها را همیشه جدا از هم نگه دارند و انتقال این اقلام از فضای کثیف به فضای تمیز مجاز نمیباشد. با جداسازی فیزیکی اتاقهای تمیز و کثیف از همدیگر و انجام فعالیتهای قبل و بعد از PCR در فضاهای جداگانه آلودگی به میزان قابل توجهی کاهش مییابد. بنابراین در هنگام طراحی آزمایشگاه مولکولی ، حداقل دو فضای مجزا باید در نظر گرفته شود. اتاقهای تمیز و کثیف به ترتیب Pre-PCR و Post-PCR گفته میشوند. با این حال در صورت وجود فضای کافی، برای ایجاد یک آزمایشگاه مولکولی ایدهآل بهتر است چهار اتاق مجزا برای آمادهسازی معرفها، آمادهسازی نمونه، مرحله PCR و مراحل پس از PCR در نظر گرفته شود. هر اتاق باید تجهیزات لباس و مواد مصرفی خود را داشته باشد و نباید مواد و تجهیزات بین اتاقها جابهجا شود.
گردش کار در آزمایشگاههای مولکولی باید یکطرفه و از سمت تمیز به سمت کثیف باشد و هیچ ماده ای ازسمت کثیف نباید به سمت تمیز آورده شود. در صورتی که همه مراحل به ناچار باید در یک اتاق انجام شود، برای هر کاری باید هودها و فضاهای جداگانه در نظر گرفت و مراحل قبل و بعد از PCR را در ساعتهای مختلف روز انجام داد. بعد از انجام هرکاری باید از نور UV برای استریل کردن فضاها استفاده شود.
طراحی و گردش کار در یک آزمایشگاه مولکولی بصورت زیر می باشد.
فشار هوا در هر آزمایشگاه باید جداگانه تنظیم شود و هر فضایی باید تهویه مخصوص خود را داشته باشد. گردش هوا بین آزمایشگاههای قبل و بعد از PCR یکی از مهمترین منابع آلودگی در آزمایشگاههای مولکولی است. در فشار هوای مثبت، فشار هوای داخل اتاق بیشتر از فشار هوای بیرون اتاق است و از ورود مواد ناخواسته به داخل اتاق جلوگیری میشود. در اتاق قبل از PCR فشار هوا باید مثبت باشد تا از ورود هوای آلوده به داخل اتاق جلوگیری شود . از طرف دیگر فشار اتاق بعد از PCR باید منفی باشد تا هوا را در داخل خود نگه دارد و از خروج امپلیکونها جلوگیری شود. درها باید بسته نگه داشته شوند تا فشار مثبت و منفی اتاقها حفظ شود. اتاقهای قبل و بعد از PCR باید کانال تهویه جداگانه داشته باشند و در جهتهای مختلفی باز شوند.
اشعه UV باعث آسیب DNA میشود و برای از بین بردن آلودگیهای DNA میتواند مفید باشد. DNA درحالت خشک در برابر UV مقاومتر از DNA محلول در آب میباشد و بنابراین استفاده از UV برای حذف آلودگی از سطوح خشک آزمایشگاهی اثر کمتری دارد. منبع نور UV را میتوان روی سقف یا روی میز آزمایشگاه قرار داد و اُزن تولید شده ناشی از تابش اشعه ماوراء بنفش باید تهویه گردد. در نتیجه اکسیداسیون، رسوباتی روی شیشه لامپ جمع میشود که اثربخشی UV را کاهش میدهد و این رسوبات ماهانه باید حذف شوند.
تستهای مولکولی در اغلب موارد کاملاً حساس و اختصاصی هستند و نتایج بسیار دقیقی را در پی خواهند داشت. مکانیسمهای کنترلی متعددی مانند تایید توالی پرایمر و پروب، بهینه بودن شرایط انجام آزمایش و استفاده از کنترلهای منفی برای کاهش نتایج مثبت کاذب باید اعمال شود. روشهای تشخیصی فعلی بسیار حساس بوده و امکان تشخیص و تکثیر یک مولکول DNA را دارند. بنابراین امکان تکثیر و تشخیص آلودگیهای DNA را باید مد نظر قرار داد و استراتژی پیشگیری از آلودگی باید در اولویت آزمایشگاههای مولکولی قرار گیرد.
آلودگی متقاطع (Cross-Contamination) یکی از منابع خطا و آلودگی در آزمایشگاههای مولکولی است و ممکن است در هر مرحله از فرآیند پردازش نمونه یا در حین استخراج DNA رخ دهد و در نتیجه باعث نتایج مثبت یا منفی کاذب میشود. میکروارگانیسمها (ویروسها، باکتریها و..) نیز ممکن است در طی این فرآیندها از یک مورد به مورد دیگر منتقل شوند.
DNA تکثیر شده از واکنشهای قبلی، کنترلهای مثبت مورد استفاده در آزمایشات، نمونههای حاوی پلاسمید، لباسها، زبالههای آزمایشگاهی و سطوح تمیز نشده، از منابع بالقوه آلودگی در آزمایشگاههای مولکولی هستند. به منظور پیشگیری از آلودگی و کنترل آن، شرایط فیزیکی مناسب، طراحی فضاها، بکارگیری دقیق تکنیکهای آزمایشگاهی، پروتکلهای کنترل محیطی و برنامه گردش کار ضروری است و فقط پرسنل مسئول و آموزش دیده باید در محیط آزمایشگاه حضور داشته باشند.
مدیریت کیفیت در تمام مراحل ارزیابی آزمایشگاه تشخیص مولکولی ضروری است و یک جزء مهم در انجام آزمایشات مولکولی محسوب میشود.
در صورتی که قصد راهاندازی آزمایشگاه مولکولی ژنتیک و تشخیص بیماریهای عفونی را دارید با کارشناسان شرکت نماژنآزما تماس بگیرید.
فنآوریهای تشخیصی به سرعت در حال پیشرفت هستند و باید پذیرفت که در مراحل اولیه درک استفاده از مفاهیم پیچیده ژنومیکس و پروتئومیکس هستیم و بسیاری از وعدههای تشخیص مولکولی هنوز محقق نشدهاند. شناسایی نشانگرهای مولکولی در جمعیتهای مختلف ، شناسایی سریع پاتوژنها، مولتیپلکس کردن آزمایشات، توالی یابی ژنتیکی، ادغام روشهای ژنتیکی با فنآوری IT و روشهای تحلیل دادهها از چالشهای پیشرو است. انتظار میرود تلفیق فنآوریهای جدید مانند نانو با تکنینکهای تشخیصی فعلی، کارایی بیشتری را در پی داشته باشد. این فنآوری میتواند علاوه بر افزایش حساسیت و اختصاصیت تستهای تشخیصی امکان مولتیپلکس کردن را نیز فراهم کند. توالییابی مبتنی بر نانو از دیگر رویکردهایی است که مورد توجه پژوهشگران قرار دارد. در نهایت پیشرفتها به سمتی باید بروند که این دسته از تستهای تشخیصی در آزمایشگاههای کمتر تخصص یافته نیز به آسانی قابل انجام باشند و زمان انجام آزمایشات نیز کاهش یابد.
بیماری آلزایمر شایعترین نوع زوال عقل است. این بیماری، یک بیماری پیشرونده است که با از دست دادن خفیف حافظه، شروع میشود و احتمالاً منجر به از دست دادن توانایی انجام مکالمه و واکنش به محیط میشود. بیماری آلزایمر قسمتهایی از مغز را درگیر میکند که فکر، حافظه و زبان را کنترل میکند.
تا امروز پیشگیری و درمان موثری برای بیماری آلزایمر ارائه نشدهاست. بنابراین توجه به تغذیه در این شرایط اهمیت ویژهای دارد. این مداخلات پیشگیرانه زمانی اثر بخش خواهد بود که علائم بیماری شروع نشده باشد، مثلا حدود پنجاه سالگی. مطالعات نشان دادهاند که عادات غذایی که منجر به بیماریهای قلبی عروقی میشوند، شانس ابتلا به آلزایمر را نیز افزایش میدهند. از سوی دیگر رژیم غذایی غنی از آنتیاکسیدان، فیبر، امگا3 و اسیدهای چرب غیر اشباع احتمالاً اثر محافظتی بر روند تخریب عصبی دارند. اثر مفید برخی از مواد غذایی مانند گلوتاتیون، پلیفنلها ، کورکورمین، کوآنزیم Q10 ، ویتامینهای B6، B12، اسید فولیک، اسیدهای چرب غیراشباع ، لسیتین، کافئین و برخی باکتریهای پروبیوتیک بر بیماری آلزایمر نشان داده شده است. از طرف دیگر رژیم غذایی حاوی اسید های چرب اشباع و اسیدهای چرب شاخهدار باعث پیشرفت آلزایمر میگردند.

سابقه خانوادگی برای ابتلای فرد به آلزایمر ضروری نیست. در واقع آلزایمر یک بیماری وابسته به سـن است. با این حال، تحقیقات نشان میدهد کسانی که والدین، خواهر و یا برادرشان مبتلا به آلزایمر هستند، نسبت به کسانی که بستگان درجه یک مبتلا به آلزایمر ندارند، بیشتر در معرض ابتلا به این بیماری قرار دارند. کسانی که بیش از یک خویشاوند درجه یک مبتلا به آلزایمر دارند در معرض خطر بیشتری هستند. هنگامی که بیماریهایی مانند آلزایمر و سایر بیماریهای مرتبط با زوال عقل در خانواده ها ایجاد میشوند، ژنتیک (عوامل ارثی)، عوامل محیطی – یا هر دو – ممکن است نقش داشته باشند.
دو دسته از ژنها وجود دارند که بر ابتلای فرد به بیماری تأثیر میگذارند: (1) ژن های مستعدکننده و (2) ژنهای تعییـن کننده . محققان ژنهای ارثی آلزایمر را در هر دو دسته شناسایی کردهاند.
– تاکنون، محققان بیش از 20 ژن را یافتهاند که ممکن است خطر ابتلا به بیماری آلزایمر را افزایش دهد. ژنهای خطر یا مستعدکننده احتمال ابتلا به بیماری را افزایش میدهند، اما تضمین نمیکنند که این بیماری رخ دهد. APOE-e4 اولین ژن خطر شناسایی شدهاست و همچنان به عنوان موثرترین ژن در افزایش احتمال بروز آلزایمر تلقی می¬شود. محققان تخمین میزنند که بین 40 تا 65 درصد از افراد مبتلا به آلزایمر دارای ژن APOE-e4 هستند.
APOE-e4 یکی از سه شکل رایج ژن APOE است. بقیهی آنها شامل APOE-e2 و APOE-e3 هستند. همهی ما یک کپی از نوعی APOE را از هر یک از والدین به ارث میبریم. کسانی که یک کپی از APOE-e4 را از مادر یا پدر خود به ارث میبرند، در معرض خطر ابتلا به آلزایمر هستند. کسانی که دو نسخه از آن را از مادر و پدر خود به ارث میبرند، احتمال بروز آلزایمر در آنها بیشتر است، اما حتمی نیست. علاوه بر افزایش خطر، APOE-e4 ممکن است سبب شود تا علائم بیماری در سنین پایینتر از حد معمول بروز نماید. تخمین زده میشود 20-30٪ از افراد در ایالات متحده یک یا دو نسخه از APOE-e4 را دارند؛ از این میان، تقریباً 2٪ هر دو نسخه از APOE-e4 را در ژنوم خود دارند.
2- ژنهای قطعی (Deterministic Genes)، مستقیماً باعث ایجاد بیماری میشوند و وجود آنها تضمینی است بر اینکه فرد حامل، دچار Alzheimer’s disease میشود. این گروه از ژنها سبب ایجاد فرم وراثتی آلزایمر هستند. البته فقط در مواردی نادر، بیماری آلزایمر، ارثی یا خانوادگی است که کمتر از پنج درصد از همهی موارد این بیماری را تشکیل میدهد. تاکنون سه ژن عامل فرم خانوادگی بیماری آلزایمر، کشف شده است: دو ژن پرسنیلین (PSEN1) و (PSEN2) و یک ژن پروتئین پیشساز آمیلوئید .(APP) این سه ژن در مجموع مسئول حدود نیمی از موارد خانوادگی آلزایمر هستند. اگر والدین هر یک از ژنهای معیوب (PSEN1، PSEN2 یا APP) را داشته باشند، فرزندانشان به احتمال 50 درصد این بیماری را به ارث خواهند برد. اگر فردی ژن عامل بیماری را به ارث برده باشد، به طور قطع میتوان گفت که بین اوایل دهه 40 تا اواسط 50 سالگی به بیماری آلزایمر مبتلا خواهد شد. تغییرات در این ژنها به ندرت باعث ایجاد بیماری آلزایمر در افراد 65 سال و بالاتر میشود.
در بیشتر موارد، بیماری آلزایمر فرم تک گیر (Sporadic) است. به این مفهوم که فرم وراثتی ندارند و پیشتر در خانواده دیده نشده است. بیماری آلزایمر به دلیل ترکیب پیچیدهای از ژنها، محیط و سبک زندگی بروز می¬کند. هیچ آزمایش ژنتیکی قابل اعتمادی، برای تشخیص فرمSporadic آلزایمر وجود ندارد؛ زیرا در بهترین حالت، فقط میتواند به میزان حساسیت فرد نسبت به بیماری اشاره کند. این آزمایش هرگز نمیتواند پیشبینی کند که آیا فرد به بیماری آلزایمر مبتلا میشود یا خیر. بنابراین، در اکثر موارد، آزمایش ژنتیک برای این بیماری توصیه نمیشود.
اکثریت قریب به اتفاق افراد مبتلا به آلزایمر، مبتلا به آلزایمر دیررس هستند که در سن 65 سالگی یا بالاتر رخ میدهد.
اگرچه ژنهای ارثی که باعث “آلزایمر خانوادگی” میشوند نادر هستند، اما کشف آنها سرنخهای مهمی را ارائه کردهاست، که به درک ما از آلزایمر کمک میکند. همه این ژنها بر پردازش یا تولید قطعه پروتئینی بتا آمیلوئید، جزء اصلی پلاکهای عامل آلزایمر، تأثیر میگذارند. بتا آمیلوئید مظنون اصلی کاهش و مرگ سلولهای مغزی است. آدوکانوماب(Aducanumab) یا (Aduhelm™)، دارویی تایید شده توسط FDA، اولین درمانی است که نشان میدهد حذف آمیلوئید از مغز احتمالاً کاهش عملکرد شناختی و عملکردی را در افراد مبتلا به آلزایمر اولیه کاهش میدهد. چندین درمان دیگر برای هدف قرار دادن آمیلوئید نیز در حال توسعه هستند.
دو تحقیق بینالمللی برای به دست آوردن بینش بیشتر در مورد بیماری آلزایمر با مطالعه افراد دارای ژنهای قطعی آلزایمر در حال انجام است:
(1) شبکه آلزایمر ارثی غالب (DIAN: Dominantly Inherited Alzheimer Network)، که توسط موسسه ملی کهنسالی (NIA) حمایت میشود، و شامل 10 مرکز تحقیقاتی برجسته در ایالات متحده، بریتانیا و استرالیا میباشد.
(2) ابتکار پیشگیری از آلزایمر (API: Alzheimer’s Prevention Initiative) که بر یک خانواده گسترده در آنتیوکیا (Antioquia) کلمبیا واقع در آمریکای جنوبی متمرکز است. این خانواده با 5000 عضو، بزرگترین خانواده جهان است که ژن عامل آلزایمر در آن شناسایی شده است.
آزمایشهای ژنتیکی هم برای APOE-e4 و هم برای ژنهای نادری که مستقیماً باعث آلزایمر میشوند در دسترس است. ولی انجمن آلزایمر نسبت به انجـام آزمایشهای ژنتیکی معمول برای بررسی خطر ابتلا به آلزایمر این هشدار را میدهد که فرد قبل از انجـام آزمایش، مشاوره مناسب را دریافت کرده و اطلاعات لازم برای تصمیمگیری آگاهانه، از جمله عوامل اجتماعی و اقتصادی را که میتواند با داشتن این اطلاعات ژنتیکی تحت تأثیر قرار گیرد، درک کند. با این حال، ممکن است موارد خاصی وجود داشته باشد که فردی که مبتلا به آلزایمر است باید آزمایش ژنتیک را با پزشک خود در میان بگذارد، زیرا نتایج میتواند بر تصمیم درمانی تأثیر بگذارد. به عنوان مثال، افرادی که واجد شرایط استفاده از درمانهای ضد آمیلوئید مانند آدوکانوماب هستند، در صورت داشتن ژن APOE-e4 ممکن است در معرض خطر یک عارضه جانبی جدی باشند. افراد باید قبل و بعد از تصمیمگیری برای انجام آزمایش، به دنبال خدمات یک مشاور ژنتیک باشند.
تمام 30000 ژنی که نقشه بیولوژیکی یک انسان را کد میکنند، روی 23 جفت کروموزوم قرار دارند. تصویر زیر، کروموزومها را نشان میدهد و ژنهای APP، PS-1، PS-2و APOE4 به ترتیب روی کروموزومهای 21، 14،1 و 19 قرار دارند.
خانوادههایی که در آنها، فرد مبتلا به آلزایمر وجود دارد، همواره نگران این موضوع هستند. با این حال، توجه داشته باشید که گاهی اوقات افراد در مورد سابقه خانوادگی بیماری آلزایمر نگران هستند، در حالی که در واقع، سابقه خانوادگی متفاوتی از زوال عقل دارند.
Amyloid precursor protein (APP), discovered in 1987, is the first gene with mutations found to cause an inherited form of Alzheimer’s.